
Translate this page into:
HBG2 -158 (C>T) polymorphism and its contribution to fetal hemoglobin variability in Iraqi Kurds with beta-thalassemia minor
Address for correspondence: Prof. Nasir Al-Allawi, Department of Pathology, College of Medicine, University of Duhok, 9 Azadi Hospital Road, 1014 AM, Duhok, Iraq. E-mail: nasirallawi@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
PURPOSE:
Hemoglobin (Hb) F% is increased in up to half of beta-thalassemia (β-thal) carriers. Several polymorphisms have been linked to such variability in different populations, including HBG2 - 158(C>T) (Xmn I polymorphism) on chromosome 11. To determine the role of this polymorphism in such variability among Iraqi Kurds, the current study was initiated.
MATERIALS AND METHODS:
A total of 102 consecutive patients diagnosed as β-thal minor were enrolled. The enrollees had their diagnosis based on peripheral blood counts and high-performance liquid chromatography to determine HbA2 and HbF. All enrollees had their DNA extracted by phenol-chloroform method and Xmn I polymorphism detected by restriction fragment length polymorphism-polymerase chain reaction.
RESULTS:
The mean age (standard deviation [SD]) of the 102 enrollees was 25.4 (14.0) years, and the enrollees included 48 males and 54 females. Xmn I polymorphism was identified in heterozygous state in 46 (45.1%) patients and in homozygous state in one patient (0.98%). Thus, the minor allele frequency of this polymorphism was 0.235 in the studied group. There were no significant differences in red cell indices and HbA2% in carriers of the minor allele compared to noncarriers, while HbF% and absolute HbF concentrations were significantly higher in the former subgroup (P = 0.032 and 0.014, respectively). This polymorphism's contribution to HbF variability was found to be 5.8% in the studied sample. Furthermore, those with HbF ≥2% were 3.2 folds more likely to carry the minor allele.
CONCLUSIONS:
Xmn I polymorphism is frequently encountered in Iraqi Kurds with β-thal minor, and it is significantly associated with higher fetal hemoglobin in these patients.
Keywords
Beta-thalassemia
Iraq
Kurds
rs7482144
Xmn I polymorphism

Introduction
Beta-thalassemia (β-thal) is an autosomal recessive inherited disorder of hemoglobin (Hb) synthesis, associated with a defect in the synthesis of β-globin chains.[1] Its inheritance is associated with a variety of phenotypes ranging from severe transfusion-dependent thalassemia major to usually asymptomatic thalassemia minor, with an intermedia phenotype in between.[2] The major phenotype is due to homozygous or compound heterozygous β-thal gene inheritance, while the minor is heterozygous for the mutant allele. The intermedia phenotype genetics are much more complex.[3] In addition to the type of β-thal mutation, other modulators are responsible for the variability in phenotype in this inherited disorder. One such modulator is inheritance of determinants associated with increased γ chain production, with resultant increase in HbF leading to reduction in α:β ratio.[4] There are three major quantitative trait loci (QTLs) on chromosomes 11, 6, and 2 related to this γ chain modulation. One of the most significant single-nucleotide polymorphisms relevant to these QTLs is located on the HBG2 locus on chromosome 11 (HBG2 - 158 [C>T] [rs7482144]). Its minor allele creates a site for the restriction endonuclease Xmn I and hence the name Xmn I polymorphism.[4567]
While earlier studies have focused and documented the contribution of Xmn I polymorphism to phenotype and HbF levels in homozygous and compound heterozygous β-thal in various populations including Iraq,[6891011] such contribution particularly to HbF has been subject to controversy in heterozygous β-thal (thal minor)[12131415] and was not addressed in Iraqi patients, and that is why this study was initiated.
Materials and Methods
A total of 102 consecutive patients (aged 2 years or older) diagnosed as β-thal minor by two specialist laboratories in Duhok, Kurdistan, Iraq, were recruited. All enrollees had a full blood count and red cell indices determined using a Hematology Analyzer (Sysmex XP-300, USA). This instrument is calibrated daily by calibrators provided by the manufacturers. Quantitation of HbF and HbA2 and exclusion of other hemoglobinopathies were performed by high-performance liquid chromatography using D-10 short thalassemia program (Bio-Rad Laboratories Inc., CA, USA). Thereafter, patients had their DNA extracted by a phenol-chloroform method.[16] The extracted DNA was then amplified using an AB2720 Thermocycler (Applied Biosystems, USA) for a 650 bp sequence in the promoter region of the Gγ-globin gene. The primers used were as follows: Forward 5’ AAC TGT TGC TTT ATA GGA TTT T3’ and Reverse 5’ AGG AGC TTA TTG ATA ACT CAG AC 3’. The polymerase chain reaction (PCR) program consisted of pre-PCR denaturation at 94°C for 2 min, followed by 30 cycles of denaturation at 95°C 1 min, annealing 60°C 1 min, and extension 72°C 1.5 min, and post-PCR final extension for 5 min at 72°C.[16] The resultant 650 bp amplicon was digested with the enzyme Xmn I according to the manufacturer's instructions (Promega, USA), and the digestion products were run on a 2% agarose gel and visualized after ethidium bromide staining via ultraviolet transilluminator (HVD Life Sciences, Austria). This study was approved by the Ethics Committee at the College of Science, University of Duhok, Iraq, and informed consent was obtained from all participants. Statistical analysis utilized the SPSS software program (release 20, SPSS Inc., Chicago, IL, USA). Chi-square test and Student's t-test were used when applicable. To assess the effect of Xmn I polymorphism on HbF concentration variability, the latter was natural log transformed (to ensure linearity), and then, linear regression was applied. P < 0.05 was considered statistically significant.
Results
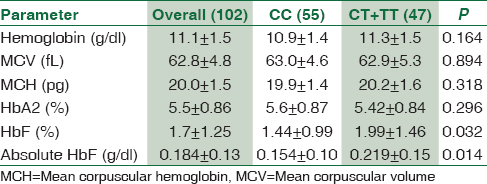
The 102 enrolled β-thal minor patients had ages ranging from 2 to 61 years, with a mean age of 25.4 ± 14.0 years, and included 48 males and 54 females. Their main hematological parameters are outlined in Table 1. HbF% varied from 0.4% to 7.7% with a mean of 1.7% ± 1.25%. In 26.5% of the enrollees, HbF% was equal or in excess of 2%.
HBG2- 158 C>T polymorphism was detected in 47/102 patients, including 46 in heterozygous state (CT) and one in homozygous state (TT). This would give a minor allele frequency (MAF) of 0.235. Figure 1 shows examples of gel electrophoresis of Xmn I digested amplicons in those with homozygous (TT) and heterozygous (CT) states for the minor allele, as well as in those with homozygous state for the wild allele (CC).
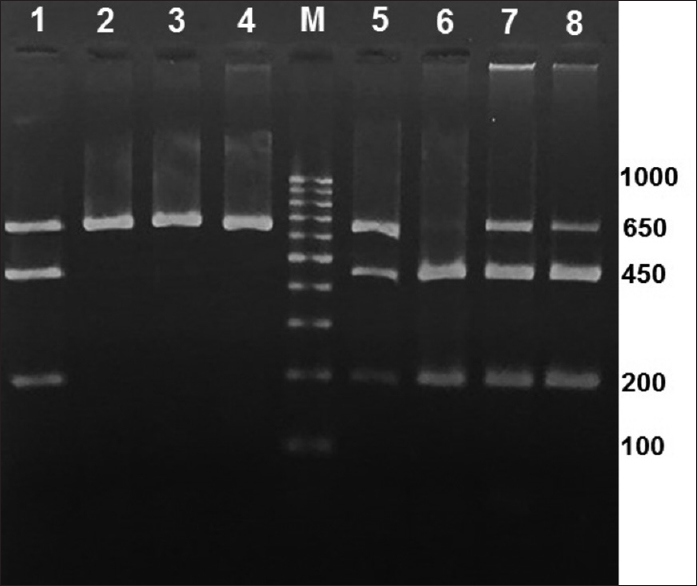
- An example of the Xmn I digested amplicons run of 2% agarose gel for HBG2-158 C>T polymorphism. Lane 1 is heterozygous control (CT) with three bands at positions 650, 450, and 200 bp. Cases 2–4 are homozygous for the wild allele (CC) with only one 650 bp band. Cases 5, 7, and 8 are heterozygous (CT), while case 6 is homozygous for the mutant allele (TT) with two bands at 450 and 200 bp
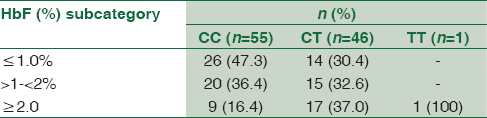
Table 1 shows a comparison of various hematological parameters between carriers (heterozygous + homozygous) of the Xmn I polymorphism and noncarriers. Clearly, there were no significant differences between any of the red cell indices, except for HbF (%) which was significantly higher in carriers of the T allele with a P = 0.032. This was even more significant when the absolute HbF concentrations were compared between carriers and noncarriers (P = 0.014). The contribution of carriage of the minor allele to HbF concentration variability was found to be a significant 5.8%, when age and sex were taken as covariates by linear regression (effect size 0.294, P = 0.016). Moreover, enrollees with HbF% ≥2% were a significant 3.2 (CI 1.2–7.6) folds more likely to be carriers of the minor allele (T) than those with lower HbF% (P = 0.012) [Table 2].
Discussion
HbF levels are variably increased in β-thal carriers as documented by many studies all over the world, with up to half of the cases having a slightly increased HbF%.[317] These increases are attributed to preferential survival of red cell precursors that synthesize relatively more γ chains. Several factors have been implicated in this increase in γ-globin chain production although twin studies have confirmed that genetic factors are the main culprit.[18] Among one of the earliest genetic factors implicated was HBG2 g.-158 C>T rs7481244 (Xmn I polymorphism), which has been reported to be associated with 3–11 folds increase in Gγ-globin chain production, by increasing the rate of the transcription of the gene, in conditions characterized by hematopoietic stress.[51920] In this series of Iraqi Kurds who are β-thal carriers, nearly 60% had increased HbF%, including 26% with a HbF in excess of 2%, which is to some extent slightly higher than many previous studies.[31317] This observation further justifies the need for addressing the issue in our population.
The MAF of HBG2 g.-158 C>T as determined in the studied sample of β-thal carriers was 0.235, which is intermediate between the rates of 0.36 in β-thal intermedia and 0.13 in β-thal major reported earlier in the same population.[9] The higher rates in thal intermedia compared to major is well documented and further supports the role of Xmn I as a modulator of disease severity in β-thal.[7921] Population studies have revealed that the MAF of Xmn I polymorphism varies between 0.10 and 0.26 in different populations.[22] Studies focusing particularly on MAF in β-thal minor, however, are not frequent. Studies on β-thal carriers from Brazil, Northern Pakistan, Turkey, and Hong Kong reported rates of 0.19, 0.16, 0.18, and 0.07, respectively.[23242526] These rates may be relevant to the underlying β-thal genotypes in these populations. Although the current study did not include molecular characterization of the underlying β-genotypes, earlier studies have documented that IVS-II-1 (G>A), codon 44 (-G), codon 5 (-CT), IVS-I-1 (G>A), and codon 39 (C>T) are the five most common β-thal mutants in carriers from our region.[27] The first two mutations have been reported as associated with Xmn I polymorphism in 89% and 75%, respectively, of carriers from Turkey,[26] while the latter two were linked to Xmn I in a lower but considerable proportion of cases in the same study. More or less similar observations were also documented by a study on Italian carriers, where Xmn I polymorphism was frequently associated with IVSII-1 and less so with codon 39 and IVS-I-1.[15] Similarly, IVS-II-1 was highly associated with Xmn I polymorphism in Greek carriers.[28] Furthermore, IVS-II-1 was quite frequently associated with Xmn I in an earlier study on thalassemia intermedia in our region.[6]
Data based on the studies on β-thal intermedia and β-thal major support a role of Xmn I polymorphism in relevance to higher HbF production and amelioration of phenotype.[9] This contribution is related to the ability to increase γ chain production in homozygous and compound heterozygous patients where there is evident erythropoietic stress. Such stress seems less evident in thalassemia carriers (heterozygous); though a mild degree of ineffective erythropoiesis, presumably due to extramedullary destruction of cells with excess alpha chains, has been documented in these carriers.[18] This may explain the significant association of this polymorphism with increased HbF in carriers, an association which is even more evident at HbF ≥2% in the current study. Similarly, several authors found a significant associated between Xmn I polymorphism and HbF levels in Chinese, Brazilian, and Portuguese β-thal carriers.[1222252930] On the other hand, an association could only be documented with the combination of Xmn I and (AT)x(T)Y polymorphisms in Italian carriers.[15] Conversely, other investigators failed to demonstrate an association between this polymorphism and HbF in carriers.[131431] The failure to document an association with HbF in the latter studies may be related to the background β-genotype or small sample size.
It is important to note that the contribution of 5.8% of this polymorphism to the HbF variability in the current study means that there is a need to study the contributions of polymorphisms in other two major QTLs, namely in BCL11A and HBS1 L/MYB. The latter has been found to contribute to variability in HbF among β-thal carriers in other populations.[1229] Other culprits that may have played a role in this variability as documented by other studies in other populations and need scrutiny are the β-genotype and alpha gene triplication.[131432]
Conclusions
It appears that Xmn I polymorphism is quite frequent in Iraqi Kurd carriers of β-thal and is associated with significantly higher HbF proportions in these carriers, though it does not explain all HbF variability and other polymorphisms related to the three major QTLs, β genotypes, and haplotypes, as well as alpha gene numbers, need to be addressed by future studies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Revisiting beta thalassemia intermedia: Past, present, and future prospects. Hematology. 2017;22:607-16.
- [Google Scholar]
- The Thalassaemia Syndromes (4th ed). Oxford: Blackwell Scientific Publications; 2001.
- Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT). Nicosia: Thalassemia International Federation; 2013. p. :3.
- Control of fetal hemoglobin: New insights emerging from genomics and clinical implications. In: Hum Mol Genet. Vol 18. 2009. p. :R216-23.
- [Google Scholar]
- β-thalassemia intermedia in Northern Iraq: A single center experience. Biomed Res Int 2014 2014:262853.
- [Google Scholar]
- Genetic modifiers in β-thalassemia intermedia: A study on 102 Iraqi Arab patients. Genet Test Mol Biomarkers. 2015;19:242-7.
- [Google Scholar]
- Molecular basis of β-thalassemia intermedia in Erbil province of Iraqi Kurdistan. Hemoglobin. 2015;39:178-83.
- [Google Scholar]
- Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. 2012;97:989-93.
- [Google Scholar]
- Utility of the multivariate approach in predicting β-thalassemia intermedia or β-thalassemia major types in Iranian patients. Hemoglobin. 2013;37:413-22.
- [Google Scholar]
- Polymorphic variations influencing fetal hemoglobin levels: Association study in beta-thalassemia carriers and in normal individuals of Portuguese origin. Blood Cells Mol Dis. 2015;54:315-20.
- [Google Scholar]
- Molecular studies of beta-thalassemia heterozygotes with raised Hb F levels. Hemoglobin. 2000;24:203-20.
- [Google Scholar]
- The triplicated alpha-globin gene locus in beta-thalassaemia heterozygotes: Clinical, haematological, biosynthetic and molecular studies. Br J Haematol. 1996;95:467-71.
- [Google Scholar]
- Influence of Ggamma-158C –> and beta- (AT)x(T) y globin gene polymorphisms on HbF levels in Italian beta-thalassemia carriers and wild-type subjects. Haematologica. 2006;91:1275-6.
- [Google Scholar]
- Prevention of Thalassemia and other Hemoglobin Disorders. Vol 2. Nicosia: Thalassemia International Federation; 2005.
- Prevention of Thalassaemia and other Haemoglobin Disorders. Vol 1. Nicosia: Thalassemia International Federation; 2003.
- [Google Scholar]
- Genetic influences on F cells and other hematologic variables: A twin heritability study. Blood. 2000;95:342-6.
- [Google Scholar]
- DNA sequence variation associated with elevated fetal G gamma globin production. Blood. 1985;66:783-7.
- [Google Scholar]
- Variation of HbF and F-cell number with the G-gamma xmn I (C-T) polymorphism in normal individuals. Blood. 1992;79:832-3.
- [Google Scholar]
- Factors regulating Hb F synthesis in thalassemic diseases. BMC Blood Disord. 2002;2:2.
- [Google Scholar]
- XmnI polymorphism frequency in heterozygote beta thalassemia subjects and its relation to fetal hemoglobin levels. Rev Bras Hematol Hemoter. 2011;33:483.
- [Google Scholar]
- Frequency of gγ-globin promoter -158 (C>T) xmnI polymorphism in patients with homozygous/compound heterozygous beta thalassaemia. Hematol Oncol Stem Cell Ther. 2015;8:10-5.
- [Google Scholar]
- Variation and heritability of Hb F and F-cells among beta-thalassemia heterozygotes in Hong Kong. Am J Hematol. 2008;83:458-64.
- [Google Scholar]
- Frequency of Gγ-globin promoter -158 (C>T) XmnI polymorphism in Denizli, Turkey. Int J Phys Sci. 2012;7:1927-31.
- [Google Scholar]
- Molecular characterization of beta-thalassemia in the Dohuk region of Iraq. Hemoglobin. 2006;30:479-86.
- [Google Scholar]
- HbF production in beta thalassemia heterozygotes for IVS-II-1 G → A beta (0) –globin mutation.Implications of the haplotype and Gγ -158 C → T mutation on Hb F level. Am J Hematol. 2000;64:151-5.
- [Google Scholar]
- A multi-locus approach to characterization of major quantitative trait loci influencing Hb F regulation in Chinese β-thalassemia carriers. Hemoglobin. 2016;40:400-4.
- [Google Scholar]
- Beta thalassemia mutations and single nucleotide polymorphism at -158 of the gamma-globin gene associated with altered levels of HbF in beta thalassemia heterozygotes. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21:498-501.
- [Google Scholar]
- The triplicated alpha gene locus and beta thalassaemia. Br J Haematol. 1983;54:201-7.
- [Google Scholar]
- Fetal hemoglobin in normal adults and beta-thalassemia heterozygotes. Hum Genet. 1990;85:106-10.
- [Google Scholar]



