
Translate this page into:
Hemoglobin E Hemoglobinopathy in an Adult from Assam with Unusual Presentation: A Diagnostic Dilemma
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hemoglobin E (HbE) is estimated to affect at least one million people around the world. Carrier frequency of hemoglobin E/β-thalassemia (HbE/β-thalassemia) is highest in Southeast Asia, reaching as high as 60% in parts of Thailand, Laos, and Cambodia. In the Indian subcontinent, highest frequency is observed in The Northeast regions, but relatively rare in rest of the country. Increasing migration of population from highly affected areas is resulting in rising prevalence in The South and other parts of India. HbE/β-thalassemia is characterized by marked clinical diversity, phenotypic instability, and age-related changes in adaptation to anemia. This paper reports a case of HbE disease in an adult immigrant from Assam and documents the difficulties encountered in the definitive subtyping of HbE hemoglobinopathy. Distinguishing between homozygous HbE disease and HbE/β-thalassemia is a challenge to hematopathologist as both are clinically and hematologically similar.
Keywords
Adaptation to anemia
clinical diversity
hemoglobin E/β-thalassemia
phenotypic instability

INTRODUCTION
Thalassemias are worldwide genetic disorders characterized by defective globin chain synthesis as a consequence of a large number of different genetic lesions. Hemoglobin E (HbE) (α2β226glu-lys) is the most common Hb variant in Southeast Asia and the second most prevalent globally.[123] HbE results from a glutamate to lysine substitution in codon #26 of the β globin gene, which produces structurally abnormal Hb. As well, Hb is synthesized at a reduced rate and behaves like a mild form of β-thalassemia.[1245] Hemoglobin E/β-thalassemia (HbE/β-thalassemia) is responsible for approximately one-half of all severe β-thalassemias globally.[4] Its phenotypic variability and instability and the limited understanding of its natural history, combine to make the management of HbE/β-thalassemia particularly challenging.
In the Indian subcontinent, HbE is mostly restricted to North-Eastern states, i.e., West Bengal, Assam, Andhra Pradesh, Nagaland, Manipur, Tripura, and Meghalaya with an average allele frequency of 10.9%. While previously rarely diagnosed in South India, increasing migration of population from highly affected areas is resulting in rising prevalence in South and other parts of India. Identification of this Hb variant is important because patients with doubly heterozygous HbE/β-thalassemia may present clinically as thalassemia major and if not treated can lead to dreadful complications.[26]
HbE/β-thalassemia is responsible for approximately one-half of all severe β-thalassemias worldwide, carrier frequency being highest in Southeast Asia and fall across the Indian subcontinent. Hb E/β-thalassemia is detected by means of newborn screening programs in highly prevalent regions.[24789]
Capillary electrophoresis is capable of distinguishing HbE from HbA2, thus permitting quantification of HbA2 in patients with HbE. Mean HbA2 in HbE trait is 3.4% and in HbE homozygotes is 4.4%. Measurement of HbA2 in the presence of HbE by capillary electrophoresis is advantageous because heterozygotes with HbA2 outside the range 3–4% ± 0.4% can prompt evaluation for additional abnormalities in Hb production, especially α-or β-thalassemia. HbE homozygous patients present with HbE values between 70% and 90%. While HbE levels are usually <40% in HbE heterozygotes, in double heterozygous HbE/β-thalassemia its level falls in the range of 40–75%. HbE/β-thalassemia typically has a greater degree of HbF elevation ranging from 14% to 40%. Capillary electrophoresis in HbE/β0-thalassemia shows bands in HbE, HbA2, and HbF regions. HbE/β+-thalassemia shows bands in HbE, HbA, HbA2, and HbF regions.[2510]
The main aim of this report is to increase the awareness of this relatively rare disorder. Identification of this Hb variant is important because patients with doubly heterozygous HbE/β-thalassemia may present clinically as thalassemia major. The consequences of HbE/β-thalassemia when not treated can be heart failure, hepatosplenomegaly, postsplenectomy thrombotic complications, and iron overload.
CASE REPORT
A 30-year-old Assami male, a migrant from Assam, presented with a history of weakness for 1 month. He had received irregular treatment for anemia on several occasions. However, he did not give a history of any previous blood transfusions. On examination, he was found to be severely pale. There were no other significant clinical findings such as organomegaly, icterus, or thalassemic bone changes. Investigations showed Hb of 4.79 gm%, red blood cell (RBC) count of 2.51 million/cmm, mean corpuscular volume of 63.46 fl, mean corpuscular hemoglobin (MCH) of 19.06 pg, MCH concentration of 30.03 gm%, and RBC distribution width of 20.7%. Peripheral smear showed anisopoikilocytic RBCs, microcytic hypochromic RBCs, target cells, and spherocytes. A Number of polychromatophils were relatively less for the degree of anemia. Not even a single reticulocyte was seen on staining with new methylene blue. Considering the peripheral smear findings, the impression of dimorphic anemia (predominantly microcytic), severe degree was rendered. Serum biochemical analysis for ferritin, Vitamin B12, and folic acid were found to be 231.70 mcg/L, 241.9 pg/mL, and 11.4 ng/mL, respectively. Sternal aspiration yielded a dry tap. One milliliter of marrow diluted with blood was aspirated from the posterior iliac crest. Bone marrow aspiration smears were hypocellular for the age and for the degree of anemia with M:E ratio of 1:1.8. He was treated for combined nutritional deficiency with Vitamin B12 and folic acid supplements. However, no improvement was seen in his clinical and hematological status. Capillary electrophoresis [Figure 1] revealed two bands corresponding to the position of HbE (93.5%) and the other at HbA2 (6.5%). Considering his history, clinical findings, and investigations, the most likely diagnosis considered was homozygous HbE disease (EE). Even though he was given blood transfusion, his condition worsened. The patient could not be followed up further as he returned back to his hometown. Since HbE/β-thalassemia cannot be entirely excluded in this case; genetic testing is of particular importance to establish the exact genetic defect causing the abnormal Hb found in this patient.
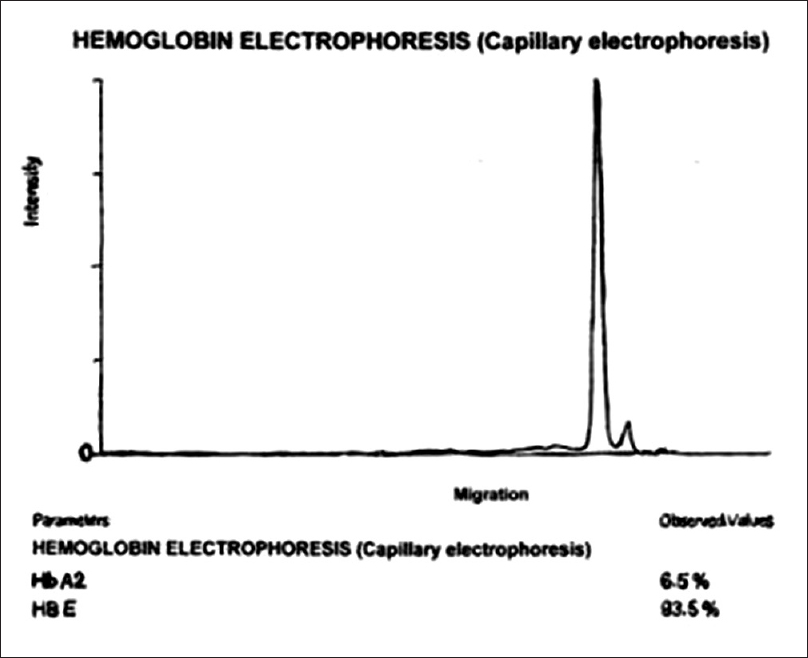
- Capillary electrophoresis
DISCUSSION
HbE gene is the mutant form of the β-globin gene. β-E chain is insufficiently produced because of a novel cryptic messenger RNA splice site, leading to thalassemic indices.[24] Since its discovery in 1954, there are uncertainties about many aspects of its pathophysiology. HbE disorders may be found in heterozygotes (AE), homozygotes (EE), and compound heterozygous states (e.g., HbE/β-thalassemia, sickle cell/HbE disease). Both heterozygotes and homozygotes are asymptomatic, minimally anemic and have microcytic and hypochromic RBCs. However, when βE allele interacts with a β-thalassemia mutation in the compound heterozygous state, a variable, and often severe anemia is produced, with Hb levels ranging from 3 to 11 g/dl.[2511]
Pathophysiology of HbE/β-thalassemia is related to many factors including reduced β chain synthesis resulting in globin chain imbalance, ineffective erythropoiesis, apoptosis, oxidative damage, and shortened red cell survival. Major genetic factors responsible for its clinical heterogeneity ranging from essentially asymptomatic to severe transfusion-dependent state includes type of β-thalassemia mutation, coinheritance of α-thalassemia, and XmnI polymorphism associated with increased synthesis of HbF. Interaction between HbE and β-thalassemia alleles is the main determinant in pathophysiology.[47812] Thai investigations suggest that patients who coinherit a mild β-thalassemia allele with HbE may have disease on mild end of the spectrum, while those who coinherit severe β+ or β0 alleles might be more severely affected.[13] In the longitudinal study of HbE/β-thalassemia in Srilanka, a positive correlation between concentration of Hb and fetal Hb, and homozygosity for XmnI polymorphism was reported.[14] Data suggest that high HbF levels in HbE/β-thalassemia result from increased erythropoietin levels leading to bone marrow expansion, and possibly increased F-cell production, giving a survival advantage to F-cells.[4141516]
HbE/β-thalassemia is characterized by widely disparate range of clinical and hematological parameters at particular stages of development. Whether this represents developmental changes in adaptation to anemia, needs to be studied. Modified “natural history” study of HbE/β-thalassemia in Sri Lankan children highlighted the instability of phenotype and a variable and changing pattern of anemia and erythroid expansion over years. In many patients, the phenotype became more stable later in development.[14] Conversely, limited data available about the clinical course of older patients indicate that many adults appear to develop worsening anemia with age. In Sri Lankan patients, a significant decline in increase in serum erythropoietin (EPO) in the presence of anemia was observed with advancing age. Increasing age is associated with a decrease of γ-globin synthesis and an increase in βE globin, both relative to α globin synthesis contributing to variation in α/βE, β/γ, and α/γ ratios. Absolute fall in HbF with advancing age is considerably amplified by thalassemia as compared to normal individuals.[414151617]
Considering his history, clinical findings, and investigations, the most likely diagnosis in our case is homozygous HbE disease. HbE disease causes only a mild, usually asymptomatic anemia with microcytosis and erythrocytosis. In our case, regular peripheral smear examination also revealed a good number of spherocytes. However, RBC count was low, and even reticulocytes were not detected on supravital staining. The presence of spherocytes could be attributed to either oxidative membrane damage or to removal of precipitated unbalanced α globin chain or removal of crystallized HbE.
Doubly heterozygous HbE/β-thalassemia is the major alternative diagnostic consideration in this case. Both HbE disease and HbE/β-thalassemia are characterized by anemia, presence of HbE by Hb electrophoresis, and target cells on peripheral blood smear. However, there can be significant differences in clinical presentation, peripheral blood smear findings, and Hb electrophoresis that may aid in distinguishing between the two. While HbE disease causes only mild asymptomatic anemia, clinically individuals with HbE/β-thalassemia can have a more devastating clinical course. Since both β genes carry a mutation in HbE disease, HbA is not seen on Hb electrophoresis. Typically, HbF is only mildly increased in this disorder. In contrast, Hb E/β-thalassemia has a greater degree of HbF elevation ranging from 13% to 40%.[10] The presence of even a minute quantity of HbA, in the absence of a history of blood transfusion, would indicate compound heterozygous HbE/β-thalassemia.
Possibility of HbE/β0-thalassemia should be strongly considered in this case because the patient presented with severe anemia (Hb of 4.7 g/dl) which is uncommon in homozygous HbE disease, and capillary electrophoresis revealed no HbA. Percentage of HbA2 outside the range 3.4% ± 0.4% prompt evaluation for additional abnormalities in Hb production, especially α-or β-thalassemia.[29] In our case, possibility of coinheritance of α-thalassemia is considered unlikely. According to a study conducted in Thailand, when percentage of HbE on Hb electrophoresis is >25%, possibility of coinheritance of α-thalassemia trait is very remote.[18]
Ineffective erythropoiesis in HbE/β-thalassemia is often coupled with dramatic expansion of hematopoietic marrow, as a result of anemic state which can lead to extensive bone deformity. In our case, the bone marrow was hypoplastic, there were no erythrocytosis and polychromatophils in peripheral blood smear. This could reflect developmental changes in adaptation to anemia. If increase in serum EPO in response to particular Hb level declines with age, drive to erythroid expansion might in parallel also decline.[481416] This suggests that recombinant EPO serves as an important therapeutic modality in treatment of HbE/β-thalassemia.
Since HbE/β-thalassemia cannot be entirely excluded in this case, genetic counseling to determine inheritance risk for future progeny is important. Hence, genetic testing or family studies, if possible, are of particular importance to establish the exact genetic defect causing abnormal Hb found in the patient. We regret that further evaluation was not possible in our case as he returned back to his hometown after his clinical condition worsened.
CONCLUSION
Compound heterozygous HbE/β-thalassemia should be considered as an alternative diagnosis in all patients with homozygous HbE disease who present with severe anemia. Age-related changes in the pattern of adaptation to anemia suggest that more cost-effective approaches to management should be explored.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Is hemoglobin instability important in the interaction between hemoglobin E and β thalassemia? Blood Am Soc Hematol. 1998;92:2141-6.
- [Google Scholar]
- The range of hemoglobin A 2 in hemoglobin E heterozygotes as determined by capillary electrophoresis. Am J Clin Pathol. 2009;132:34-8.
- [Google Scholar]
- Hemoglobin E disease in North Indian population: A report of 11 cases. Hematology. 2007;12:343-7.
- [Google Scholar]
- Hb E/beta-thalassaemia: A common & clinically diverse disorder. Indian J Med Res. 2011;134:522-31.
- [Google Scholar]
- Haemoglobin E β+-thalassaemia. A case report from Bijapur, South India. Al Ameen J Med Sci. 2009;2:82-4.
- [Google Scholar]
- Hemoglobin E disorders in Eastern Uttar Pradesh. Indian J Pathol Microbiol. 2009;52:110-2.
- [Google Scholar]
- HbE/ß-thalassemia: Basis of marked clinical diversity. Hematol Oncol Clin North Am. 2010;24:1055-70.
- [Google Scholar]
- A mechanism of ineffective erythropoiesis in ß-thalassemia/Hb E disease. Haematologica. 2010;95:716-23.
- [Google Scholar]
- Molecular and hematologic features of hemoglobin E heterozygotes with different forms of α-thalassemia in Thailand. Ann Hematol. 2003;82:612-6.
- [Google Scholar]
- Detection of Hb variants and hemoglobinopathies in Indian population using HPLC: Report of 2600 cases. Indian J Pathol Microbiol. 2010;53:57-62.
- [Google Scholar]
- Enhanced activation of autophagy in ß-thalassemia/Hb E erythroblasts during erythropoiesis. Ann Hematol. 2011;90:747-58.
- [Google Scholar]
- Severity differences in beta-thalassaemia/haemoglobin E syndromes: Implication of genetic factors. Br J Haematol. 1993;83:633-9.
- [Google Scholar]
- Intact transferrin receptors in human plasma and their relation to erythropoiesis. Blood. 1990;75:102-7.
- [Google Scholar]
- Why are hemoglobin F levels increased in HbE/ß thalassemia? Blood. 1999;94:3199-204.
- [Google Scholar]
- Molecular and hematological characterization of HbE heterozygote with alpha-thalassemia determinant. Southeast Asian J Trop Med Public Health. 1997;28(Suppl 3):100-3.
- [Google Scholar]




