
Translate this page into:
Late-onset, giant juvenile xanthogranuloma

*Corresponding author: Mona Lisa, Department of Pathology/Lab Medicine, All India Institute of Medical Sciences Deoghar, Deoghar, Jharkhand, India. monaloud@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sripati T, Lisa M, Kumar N, Roy AD, Kumar A. Late-onset, giant juvenile xanthogranuloma. J Lab Physicians. 2024;16:578-81. doi: 10.25259/JLP_44_2024
Abstract
Juvenile xanthogranuloma (JXG) is the most prevalent form of non-Langerhans cell histiocytosis. The typical presentation includes small papules or nodules in infants, often resolving spontaneously within a few years without treatment. Histologically, it showcases lipid-laden macrophages and Touton giant cells. However, atypical cases may manifest as larger lesions in older individuals, lacking foamy histiocytes and displaying increased mitotic activity. It is important to consider JXG as a potential diagnosis for skin lesions before assuming malignancy. Immunohistochemistry markers such as CD163, CD4, and CD31 can be instrumental in confirming the diagnosis. In this report, reviewing the literature, we present one such unique case of JXG.
Keywords
Juvenile xanthogranuloma
Touton giant cell
Histiocytes
Dermatopathology

INTRODUCTION
Juvenile Xanthogranuloma (JXG) is the most common form of non-Langerhans cell histiocytosis arising from the dermal dendritic cells or macrophages. Typically, it manifests as a diminutive papule or nodule in the pediatric age group, often resolving spontaneously within a few years without necessitating treatment.[1,2]
JXG belongs to a diverse group of histiocytoses, which are classified into different groups based on origin and presentation.[3] The exact cause remains unclear, but there are associations with conditions such as neurofibromatosis and leukemia, hinting at a possible link to mitogen-activated protein (MAP) kinase pathway hyperactivity.[4]
Diagnosis is confirmed by a distinct appearance of xanthomatous histiocytes in the dermis. Characteristic is the presence of Touton giant cells having a ring of nuclei surrounding a central homogeneous cytoplasm, while a foamy cytoplasm surrounds the nuclei.[1,2]
Immunohistochemistry (IHC) markers such as CD163, CD4, and CD31 help differentiate JXG from other skin lesions in cases with atypical features.[5,6]
In this study, we report such an unusual presentation with a progressively enlarging giant lesion, over 10 cm in diameter. There was an absence of foamy histiocytes, and the lesion was mitotically active. The presence of Touton giant cells and the typical IHC pro-life helped us in clenching the diagnosis.
CASE REPORT
A 22-year-old male presented with a swelling on his lower back that had been present for 10 years but recently had a sudden increase in size over the past 2 months. Upon examination, a large exophytic mass measuring approximately 12 × 10 × 4 cm was observed, with ulcerated overlying skin. A contrast-enhanced computed tomography scan revealed a large, subtly enhancing soft-tissue mass lesion in the left paramedian region at the D12-L3 vertebral level, involving the skin and subcutis. However, there was no evidence of involvement of the underlying muscle or posterior abdominal wall [Figure 1].
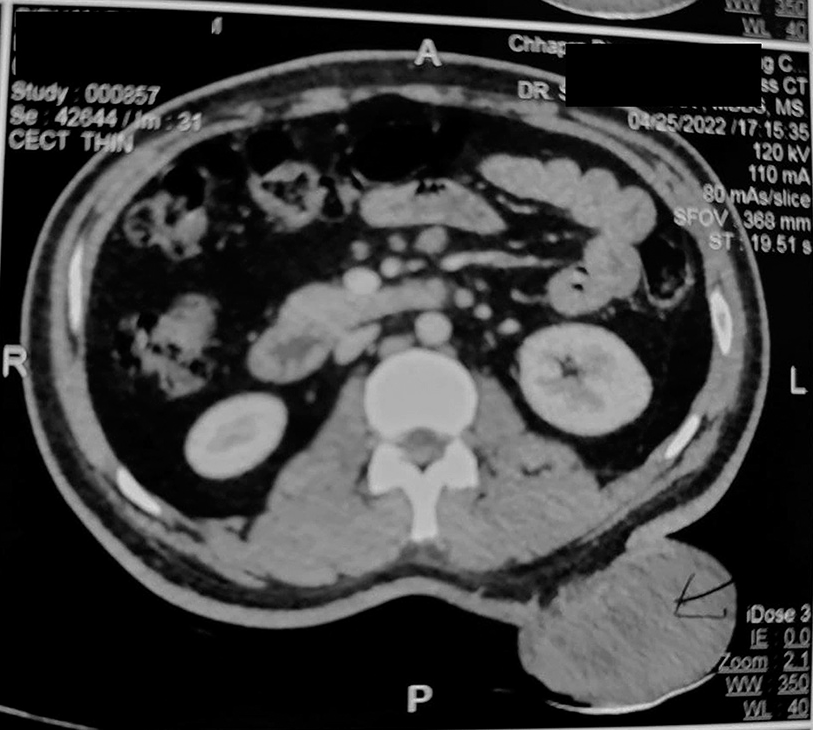
- Contrast-enhanced computed tomography (CECT) scan showed a mass at D12-L3 level, involving the overlying skin but sparing the underlying muscle and abdominal wall.
A wide local excision was performed to remove the lesion, and the resulting defect was covered with a split-thickness skin graft. The excised specimen was then sent for histopathological examination. Grossly, the specimen consisted of partially skin-covered tissue with an underlying fleshy tumor. The overlying skin exhibited ulceration, while the base of the tumor was composed of fibrofatty tissue [Figure 2a-d]. Intraoperatively, the tumor appeared well-demarcated, with no gross involvement of the underlying muscle.
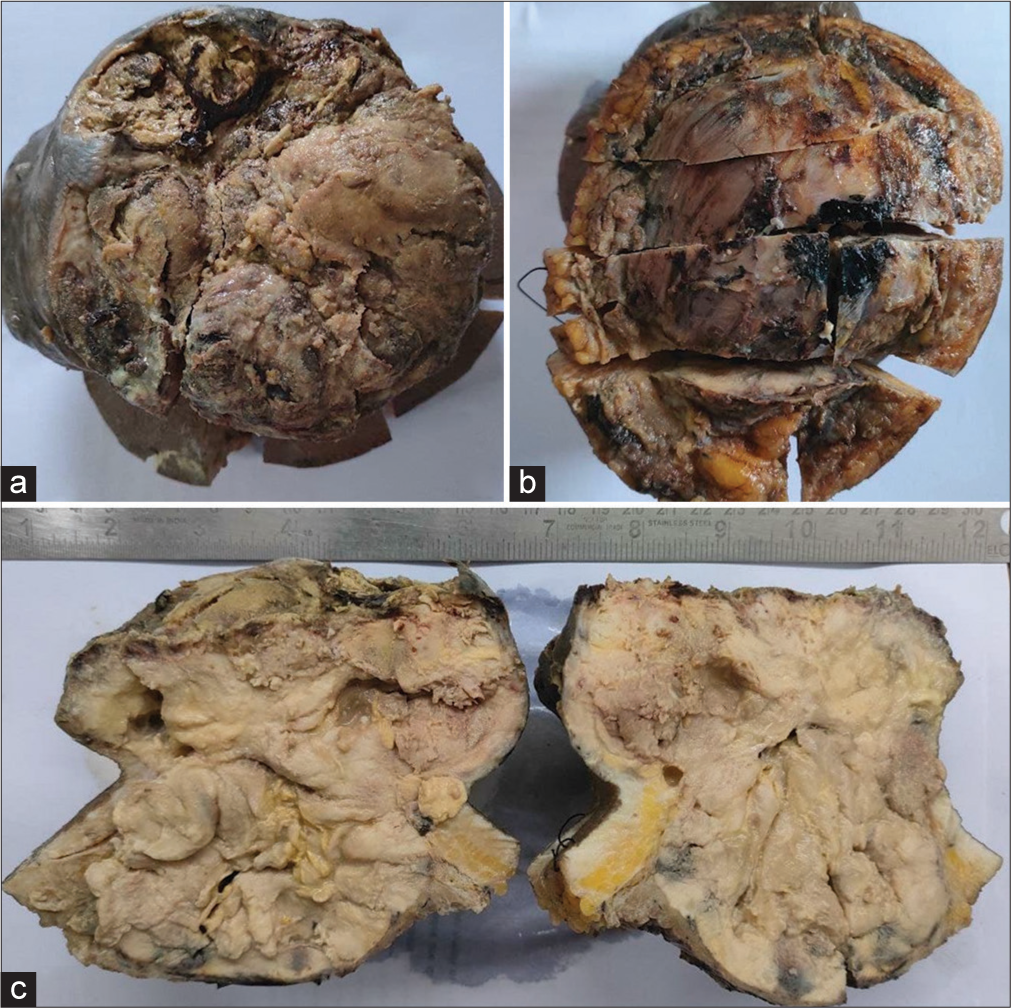
- (a) Gross picture of the lesion showing exophytic mass with ulcerated overlying skin, (b) a fibrofatty base and (c)a fleshy tumor on cut.
Microscopically, the tumor was found to be well-demarcated and non-encapsulated. There was dense infiltration of monomorphic histiocytes with abundant pink cytoplasm within the dermis, along with scattered mixed inflammatory cells, including lymphocytes, eosinophils, and plasma cells [Figure 3a]. Frequent mitotic figures were seen [Figure 3b]. Some areas of the tumor exhibited numerous Touton giant cells, and some also showed emperipolesis [Figure 3c]. Typical foamy lipid-laden histiocytes seen in JXG were infrequent. IHC revealed strong and diffuse positivity for Vimentin, CD4, and CD163 in the tumor cells [Figure 4a-c]. Weak diffuse CD31 staining and a high Ki67 proliferation index (30–40%) were also noted [Figure 4d and e ]. The tumor cells tested negative for Pan CK, TLE-1, CD45, Desmin, SMA, STAT6, S100, D2 40, CD1a, CD33, and HHV8. Based on these findings, a diagnosis of mitotically active JXG - nonlipidized subtype was made.
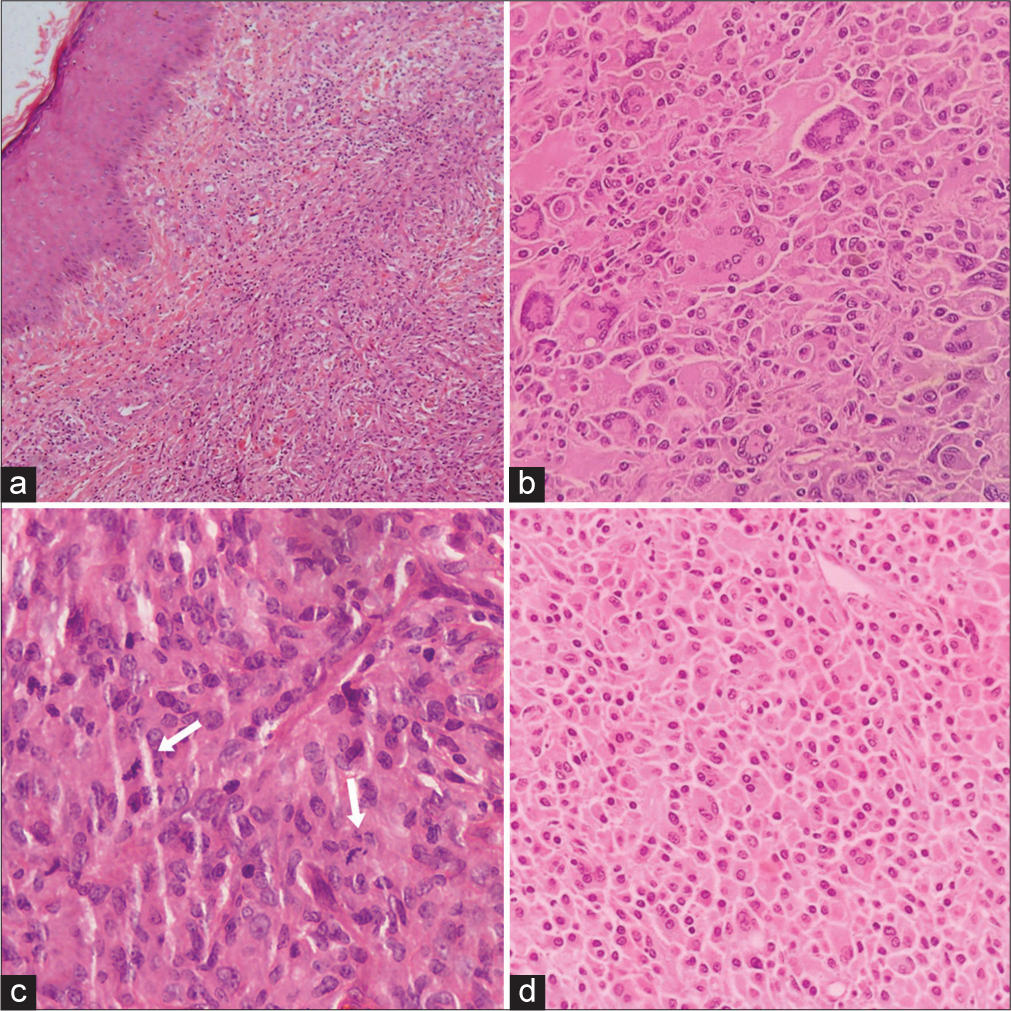
- Hematoxylin and Eosin (H&E)-stained section from the tumor in the dermis extending to subcutis. (b) Numerous Touton giant cells, histiocytes with abundant cytoplasm, some showing emperipolesis, scattered eosinophils, lymphocytes and plasma cells. (c) Mitotically active tumor (white arrows) (d) Sheets of histiocytes with abundant cytoplasm, however typical foamy cytoplasm not seen.
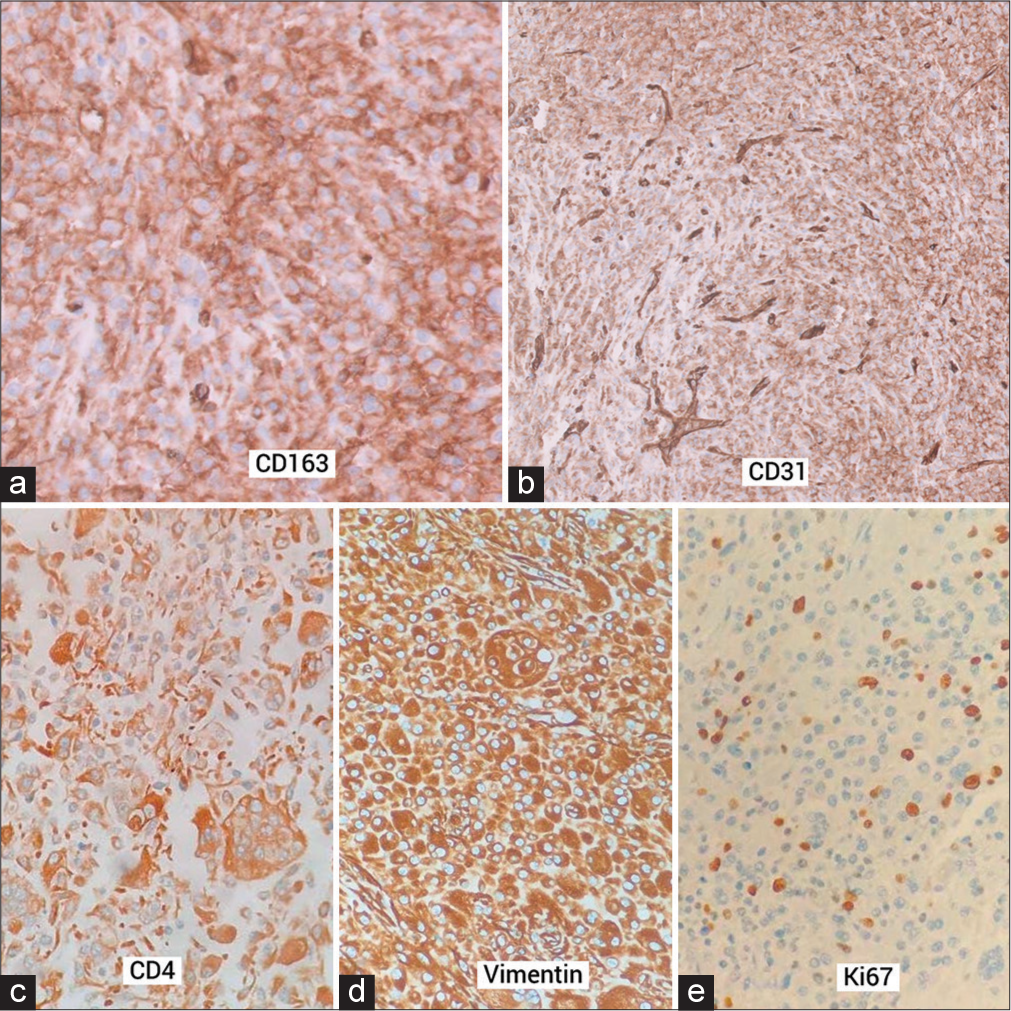
- Immunohistochemistry showed the lesional cells expressing (a) CD163, (b) weak CD31, note the blood vessels taking strong staining (c) CD4, and (d) Vimentin (e) Ki67 expression was high (30 to 40%).
A wide local excision of the tumor was already done. After this diagnosis, a thorough physical examination was done, and the computed tomography scans were re-examined to assess the presence of any systemic involvement. As no other lesion was found, so the patient was advised follow-up visits.
DISCUSSION
The histiocytoses are a heterogeneous group of proliferation of cells derived from dendritic cells or macrophages. The latest classification of these disorders consists of 5 groups, namely the Langerhans-related (L Group), cutaneous and mucocutaneous (C group), malignant histiocytoses (M group), Rosai–Dorfman disease (R group) and hemophagocytic lymphohistiocytosis and macrophage activation syndrome (H group). Solitary JXG falls under the “Group C” of histiocytoses, whereas extracutaneous and disseminated JXG is kept in the L group.[3]
The exact pathogenesis of JXG remains unknown, but there have been reported associations with other conditions, such as neurofibromatosis (types 1 and 2) and juvenile myelomonocytic leukemia. These associations suggest a possible link to MAP kinase pathway hyperactivity, which might be a shared mechanism among these hematologic disorders.[4] Regarding the cell of origin for JXG, various hypotheses have been postulated, proposing macrophages, dermal dendrocytes, dermal indeterminate cells, and CD4+ plasmacytoid monocytes as potential sources.[5,6] Further research is needed to fully understand the underlying mechanisms and origins of this intriguing condition.
Characteristically, JXG appears as a small pigmented patch or nodule and usually follows a self-limiting course, often requiring no treatment. Caputo et al. described a classification of JXG based on size, comprising small nodular or papular (2–5 mm), large nodular (5–20 mm), and giant forms (more than 20 mm).[7] Our case presented with a progressively enlarging giant lesion that measured over 10 cm in diameter.
The usual appearance – a distinct dermal infiltrate of xanthomatous histiocytes extending into the subcutis, accompanied by scattered Touton giant cells – generally confirms the diagnosis of JXG when combined with appropriate clinical context. However, atypical clinical presentation with predominantly non-lipidized histiocytes and abundant mitoses necessitates the use of IHC for confirmation, even in the presence of Touton giant cells. Touton giant cells are not pathognomonic for JXG and can occur in other xanthomatous histiocytic lesions such as Erdheim chester disease and necrobiotic xanthogranuloma.[1,2] Shapiro et al. describe a series of JXG with inconspicuous or absent foam cells and lesions showing abundant mitoses.[8] Our case also presented with abundant mitosis. Such cases should not be confused with malignant lesions. Appropriate use of IHC helps us to reach a correct diagnosis in such cases.
CD163, a marker specific to the monocyte/macrophage lineage, is distinctively expressed in JXG and not in benign fibrohistiocytic skin lesions such as dermatofibroma or benign fibrous histiocytoma.[5] Furthermore, CD4, typically found in normal plasmacytoid monocytes, is also detected in the lesional cells of JXG. In addition, CD31, a marker common in vascular-origin tumors, is expressed in the majority of JXG cases but not in dermal fibrohistiocytic tumors.[5,9]
In our case, the presence of numerous Touton giant cells alongside histiocytes expressing CD163, CD4, and CD31 conclusively confirmed the diagnosis of JXG.
CONCLUSIONS
JXG may exhibit atypical features such as a larger size, occurrence in older age groups, lack of foamy histiocytes, and numerous mitoses. It is crucial to consider JXG as a potential differential diagnosis for cutaneous lesions before considering malignant lesions. The use of a panel of IHC markers, including CD163, CD4, and CD31, proves helpful in confirming the diagnosis.
Author contribution
ML, ABR: Idea formulation; TS, AK: Final diagnosis of the case; NK, ML: Manuscript compilation.
Ethical approval
The research/study was approved at AIIMS Deoghar, approval number AIIMS/Deo/Patho/lab med/IEC/644/2024.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Juvenile xanthogranuloma: An entity with a wide clinical spectrum. Actas Dermosifiliogr (Engl Ed). 2020;111:725-33.
- [CrossRef] [PubMed] [Google Scholar]
- Juvenile xanthogranulomas in the first two decades of life: A clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol. 2003;27:579-93.
- [CrossRef] [PubMed] [Google Scholar]
- Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-81.
- [CrossRef] [PubMed] [Google Scholar]
- Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124:3007-15.
- [CrossRef] [PubMed] [Google Scholar]
- Solitary (juvenile) xanthogranuloma: A comprehensive immunohistochemical study emphasizing recently developed markers of histiocytic lineage. Hum Pathol. 2015;46:1390-7.
- [CrossRef] [PubMed] [Google Scholar]
- Uncommon histiocytic disorders: RosaiDorfman, juvenile xanthogranuloma, and Erdheim-Chester disease. Hematology Am Soc Hematol Educ Program. 2015;2015:571-8.
- [CrossRef] [PubMed] [Google Scholar]
- Unusual aspects of juvenile xanthogranuloma. J Am Acad Dermatol. 1993;29(5 Pt 2):868-70.
- [CrossRef] [PubMed] [Google Scholar]
- Juvenile xanthogranulomas with inconspicuous or absent foam cells and giant cells. J Am Acad Dermatol. 1991;24(6 Pt 1):1005-9.
- [CrossRef] [PubMed] [Google Scholar]
- "Juvenile" xanthogranuloma: An immunophenotypic study with a reappraisal of histogenesis. Am J Dermatopathol. 2001;23:104-11.
- [CrossRef] [PubMed] [Google Scholar]



