
Translate this page into:
Perinatal Autopsy Findings in a Case of De Novo Hypohidrotic Ectodermal Dysplasia
Address for correspondence: Dr. Panduranga Chikkannaiah, E-mail: pandupath@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Ectodermal dysplasia are group of inherited disorders involving the developmental defects of ectodermal structures like hair, teeth, nails, sweat glands, and others. X-linked recessive inheritance is most common. Here we describe perinatal autopsy findings in a case of de novo ectodermal dysplasia in a female fetus. To the best of our knowledge, this is the first fetal autopsy description in a case of ectodermal dysplasia.
Keywords
De novo
ectodermal dysplasia
fetal autopsy
hypohidrotic
oligodontia
trichodysplasia

INTRODUCTION
Ectodermal dysplasia (ED) are group of inherited disorders involving the developmental defects of ectodermal structure with an incidence of 1 in 1,00,000 live births.[1] This group of inheritable disorders are characterized by an abnormality of two or more structures derived from ectoderm such as the hair, teeth, nails, sweat glands, craniofacial structures, digits, and others. The X-linked recessive is the most common pattern of inheritance while autosomal dominant and recessive are rare.[23] In few cases, family history is not accountable, constituting de novo mutation.[45] There are 117 possible phenotypic types of ED have been reported.[4] Review of the literature revealed few case reports in the living individuals however to the best of our knowledge this is the first description of perinatal autopsy findings in a case of ED.
CASE REPORT
A 21-year-old primigravida with 28 weeks of gestation, presented with a history of not appreciating fetal movements since 2 days. She gave a history of nonconsanguineous marriage. Family history was not significant. There was no history of fever with rashes during pregnancy and no history of any drug intake except iron and folic acid supplements. Routine hematological and biochemical investigations were within normal limits. Sonography revealed the absence of fetal cardiac activity and intrauterine growth retardation. After obtaining consent, the pregnancy was terminated and the fetus was sent for pathological examination. On Gross examination, fetus weighed 700 g. All the fetal measurements were within normal limits. A tiny piece of umbilical cord attached to the fetus showed two vessels. There was sparse hair on scalp and all over the body. The fetus had potter's facies that is low set ears, depressed nasal bridge, and increased intercanthal distance. There was absence of lips [Figure 1a]. The nipple areola complex was absent [Figure 1b]. The nails in fingers and toes were also absent. The skin was thin and easily peelable [Figure 1b and c]. An orthopantomogram was done, which revealed decreased calcification and absence of tooth buds in many sockets (oligodontia) [Figure 1d]. No skeletal abnormalities detected on infantogram.
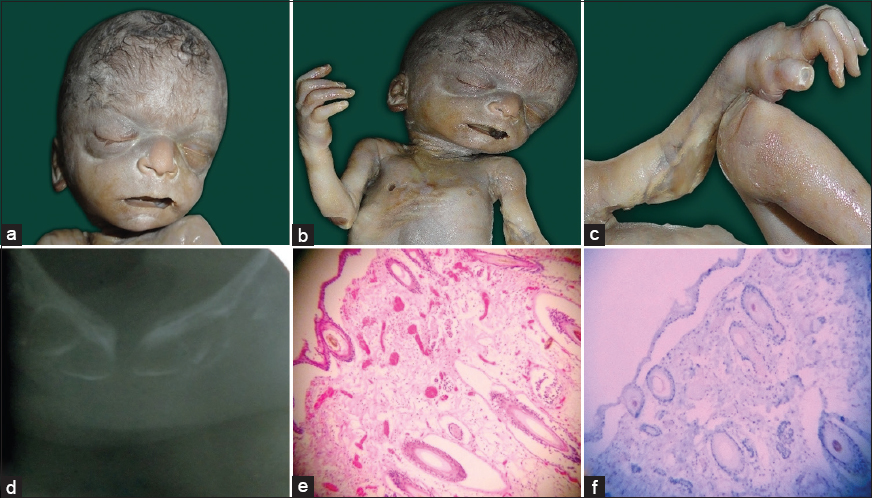
- (a) Gross photograph showing sparse hair on scalp and absence of lips, (b) Gross photograph showing sparse hair on chest, absence of nipple and skin deficient areas on left elbow joint. (c) Gross photograph showing skin deficient areas on right elbow joint and right hand. (d) Orthopantogram showing absence of tooth in the sockets. (e) Microphotograph of skin showing thinned out epidermis, immature hair follicle, and absence of sweat glands (H and E, ×10). (f) Microphotograph of skin showing negative immunostaining for herpes simplex virus (HSV) antibody (IHC HSV, ×10)
On dissection, all organs are in situs except left kidney which was malrotated and nonascended to the left lumbar region. The capsule was easily strippable and fetal lobulations were present. In the thorax, the lungs appeared hypoplastic and the heart was normal in development. Microscopic examination of the skin revealed thinned out epidermis. There was absent to scanty sebaceous glands (hypohydrosis). The hair follicles were decreased in number and were immature (hypotrichosis), the sweat glands were absent [Figure 1e]. Immunohistochemical (IHC) staining for herpes simplex virus (HSV) was carried out on skin sections and was negative [Figure 1f]. Histopathological section of the skin from age and sex matched fetus studied, which showed normal development of epidermis and adnexal structure thus confirming the nondevelopment of skin and its appendages in the present fetus. With the above features, a final diagnosis of hypohidrotic ED was made.
DISCUSSION
Ectodermal dysplasia might have been recorded as early as 1792 by Danz.[3] Weech first used the term hereditary ED to describe the congenital anomalies affecting the ectodermal structure.[6] In 1838, Wedderburn's reported ED in 10 men in a Hindu family the highlight being X-linked recessive inheritance and oligodontia in the family. Cockyane in 1933 postulated X-linked recessive inheritance in families where only males were affected and autosomal dominant if female also manifested the features. It was Levit in 1936 who pointed out that females never exhibit full syndrome, hence opening up a new possibility of de novo mutation.[45]
Ectodermal dysplasia is commonly transmitted as X-linked recessive pattern with autosomal dominant and recessive being rare.[27] The defective genes that are associated with ED are ED-1, muscle segment homeobox homology-1, ectodysplasia-1 (EDAR), and paired box gene (PAX 9).[37] Kere et al.[8] in their study, observed a new genetic locus of ED, the protein product is 135 residue transmembrane protein and is predicted to belong to noval class with a role in epithelial and mesenchymal signaling. Monreal et al.[9] in their study isolated human homolog of mouse dl (human DL) in three families, one displaying recessive inheritance and two displaying dominant inheritance. The putative protein is predicted to have single transmembrane domain and is similar to tumor necrosis factor receptor family. Congenital HSV infection can produce skin vesicle and scarring simulating ED.[10]
Organogenesis is a complex process that occurs due to synergistic interaction between ectoderm and mesoderm. ED may appear during first trimester of pregnancy. The genes ED, EDAR and others synergistically act to produce protein ectodysplasin-1, which forms a part of signaling pathway that is critical for the interaction between ectoderm and mesoderm.[4] With respect to ectodermal derived structure, 6th week of embryonic life is critical, as at this point the single layer of ectoderm divides into two cell layer leading to subsequent organ development. Dental lamina also begins at 6th week; hence, any mutation at this point leads to a severe form of ED involving all the ectodermal structure along with teeth.[1112] (as observed in our case)
Ectodermal dysplasia are primarily classified into four different types, ED1 (trichodysplasia), ED2 (dental dysplasia), ED3 (onychodysplasia), and ED4 (dyshidrosis). From these major types 150 different combinations of subtypes are classified, most common being subtype 1-2-3-4 involving all the structures that is, hypohidrotic ED. Our case also qualifies for subtype 1-2-3-4.[346]
Morphological spectrum of ED varies from case to case. Skin and teeth are most commonly involved organs. Skin is usually thin, soft and dry (as observed in our case). There is hyperkeratosis of palm and foot. There is wrinkling and pigmentation around the eyes and mouth. The lanugo hair is absent, pubic and axillary hairs are sparse. Hair over the scalp is often blond, fine, stiff, and short (as observed in our case). Eyelashes and eyelids are absent. Nails are usually normal or spoon shaped. Lacrimal gland function is impaired. In females, mammary glands are hypoplastic or aplastic and abnormalities of the nipple are also recorded. The lips are protuberant. Corneal and lenticular opacities are rare. Mental retardation has been reported in 30–50% of the patient. Dental anomalies include oligodontia of primary and permanent dentition. Complete absence of teeth is a rare event.[34613]
Other rare abnormalities recorded with ED are frontal bossing, depression of the nasal bridge, small face, central nervous system defects, and adrenal medullary defects. The pharyngeal, laryngeal, oral, and nasal mucosal glands are defective leading to dryness and repeated infections.[413] Our case in addition to classical abnormalities highlighted the absence of nipple, nails, and lips, which may be due to the severe mutation involving the genes.
It is difficult to diagnose ED in the early infancy. Classical morphological features helps in diagnosis. Testing of lacrimal gland sections, skin biopsy, sweat pore test, and molecular tests help in diagnosis of the condition in the living. Skin biopsy documents the hypohidrosis and decreased number of eccrine glands. Sweat pore test in addition to detect the affected individuals also helps in identification of carriers. In suspected cases, prenatal diagnosis is possible by chorionic villi biopsy at 10th week of gestation.[3414]
The present case had all the characteristic features of hypohidrotic ED. A near complete work up was done and HSV, which is known to cause defective embryogenesis has been ruled out. Based on morphology and histological finding we arrived at a diagnosis of hypohidrotic ED with a possibility of de novo mutation.
CONCLUSION
Ectodermal dysplasia is rare disorder and its presentation in a female fetus on autopsy makes it even rarer. Such rare case beckons a thorough evaluation with family history, radiological, and IHC methods along with genetic counseling.
ACKNOWLEDGEMENTS
We wish to acknowledge Dr. S. K. Shankar, Emeritus Professor, Dr. Anitha Mahadevan, Additional Professor and Mr. Prasanna kumar P.N Lab Technician, Human Brain Tissue Repository, Department of Neuropathology, National Institute of Mental Health and Neuroscience, Hosur Road, Bangalore - 560 029, India for helping with immunohistochemistry for HSV antibody.
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- Ectodermal dysplasia with anodontia: A report of two cases. Eur J Dent. 2010;4:215-22.
- [Google Scholar]
- Anhidrotic ectodermal dysplasia: A case report in a Nigerian child and literature review. Niger J Paediatr. 2012;39:79-83.
- [Google Scholar]
- Autosomal recessive anhidrotic ectodermal dysplasia: A rare entity. Indian J Dermatol. 2014;59:422.
- [Google Scholar]
- Gene effect in carriers of anhidrotic ectodermal dysplasia. J Med Genet. 1966;3:169-76.
- [Google Scholar]
- The ectodermal dysplasia: Severe palmoplanter hyperkeratosis and chronic angular chelitis. Indian J Dermatol. 2003;48:223-8.
- [Google Scholar]
- Genetic aspects of dental anomalies. Paediatric Dentistry Principles and Practice. (2nd ed). New Delhi: Elsevier; 2011.
- [Google Scholar]
- X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996;13:409-16.
- [Google Scholar]
- Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet. 1999;22:366-9.
- [Google Scholar]
- Herpes simplex virus infection in pregnancy. Infect Dis Obstet Gynecol 2012 2012 385697
- [Google Scholar]
- Normal skin. In: Milles SE, ed. Histology for Pathologist (3rd ed). Philadelphia: Lippincott Williams & Wilkins; 2007. p. :4-9.
- [Google Scholar]
- Develoupmental of dentition and occlusion. In: Muthu MS, Sivakumar N, eds. Paediatric Dentistry Principles and Practice (2nd ed). New Delhi: Elsevier; 2009. p. :58-62.
- [Google Scholar]
- Hypohidrotic ectodermal dysplasia. 1993-2014. GeneReviews®. Seattle, WA: University of Washington, Seattle; Available from: http://www.ncbi.nlm.nih.gov/pubmed
- [Google Scholar]



