
Translate this page into:
Primitive neuroectodermal tumor of the kidney: A rare case report
Address for correspondence: Dr. Manjari Kishore, Department of Pathology, Room No. 310, 3rd Floor, OPD Building, Dr. RML Hospital, Baba Kharak Singh Marg, New Delhi - 110 001, India. E-mail: drmanjarik@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Renal primitive neuroectodermal tumor (PNET) is a rare primary renal neoplasm. Morphologically, it may mimic small blue round-cell tumor. Hence, histopathology in conjunction with immunohistochemistry plays a significant role in correctly diagnosing this malignancy. We report a case of PNET of kidney in a 30-year-old female with an extension to inferior vena cava, who succumbed following an aggressive course of illness.
Keywords
Histopathology
immunohistochemistry
primitive neuroectodermal tumor
renal
small-round-cell tumor

Introduction
Renal primitive neuroectodermal tumor of the kidney (rPNET) is a rare malignant neoplasm to be reported in the literature.[1234] It is commonly noted in the bone and soft tissues of the body; however, rarely it can be found in other sites such as kidney, bladder, prostate, testis, ovary, and uterus.[2345] It is an aggressive tumor of young adults commonly confused with small blue round-cell tumors such as Wilms tumor, rhabdomyosarcoma, neuroblastoma, carcinoid, lymphoma, and clear-cell sarcoma of the kidney.[45] As the clinical manifestations are very much like other renal neoplasms, the diagnosis is made by histopathology in conjunction with immunohistochemical (IHC) examination. Herein, we present an interesting case of rPNET with inferior vena cava (IVC) thrombus in a young female who succumbed following fatal course of this disease.
Case Report
A 30-year-old female patient presented with complaints of right abdominal pain and significant weight loss for the past 1–1.5 months along with a progressively enlarging abdominal mass for the past 15 days. The patient had a single episode of hematuria in the past 4–5 months. On per-abdomen examination, the mass was seen to involve the right hypochondrium and right lumbar region. The mass was smooth, well defined, firm to hard, and moved with respiration.
Contrast-enhanced computed tomography (CECT) abdomen revealed a heterogeneously enhancing isodense to hypodense mass lesion arising from the right kidney replacing most of the renal parenchyma. The fat planes and other viscera such as liver, gallbladder, and suprarenal were preserved. CECT angiogram showed the same hypodense contiguous extension into the right atrium involving infra- and supra-hepatic IVC. A radical nephrectomy along with IVC and atrial thrombectomy was performed, and the formalin-fixed sample was sent to the laboratory for histopathological examination.
Grossly, the kidney was enlarged and distorted. The renal capsule and perirenal fat were intact. On serial sectioning, a tumor was seen replacing almost whole of the renal parenchyma measuring 20 cm × 14 cm × 9 cm, involving the renal pelvis and sinus [Figure 1a]. The tumor was friable, grayish-brown with areas of necrosis and hemorrhage [Figure 1b]. The right adrenals were grossly uninvolved. A gray-brown tubular muscular structure with blood clot was also identified and sectioned.
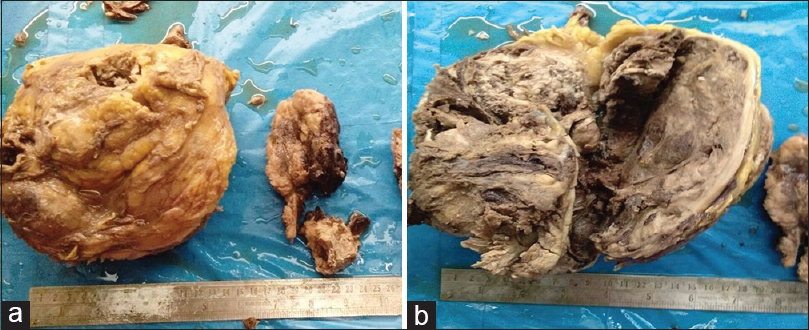
- (a and b) Grossly, the kidney was enlarged and distorted and the cut section showed a tumor replacing almost whole of the renal parenchyma measuring 20 cm × 14 cm × 9 cm, involving the renal pelvis and sinus. The tumor was friable, grayish-brown with areas of necrosis and hemorrhage
Microscopic examination showed a tumor with malignant cells arranged in solid sheets, cords, and pseudorosettes. The cells were monomorphic with small round nuclei having fine chromatin and inconspicuous nucleoli [Figure 2a–b]. Also noted were areas of normal renal parenchyma [Figure 2c] and extensive hemorrhagic areas [Figure 2d]. The tumor involved the renal parenchyma, pelvicalyceal system, and sinus with a thrombus in IVC up to right atria. However, the right adrenal and ureter were free of tumor. An IHC panel of CD99, NSE (favoring peripheral neuroectodermal tumor), WT-1 (favoring adult Wilms tumor), CD10, BCL-2, vimentin (favoring clear cell sarcoma), CD99, EMA (favoring synovial sarcoma), and CD56, synaptophysin (favoring neuroblastoma) were applied. The tumor cells showed immunoreactivity for CD99 [Figure 2e], NSE, and vimentin [Figure 2f]. Hence, based on morphology and IHC examination, a final diagnosis of PNET of the kidney (PNET) – Stage 3 was rendered.
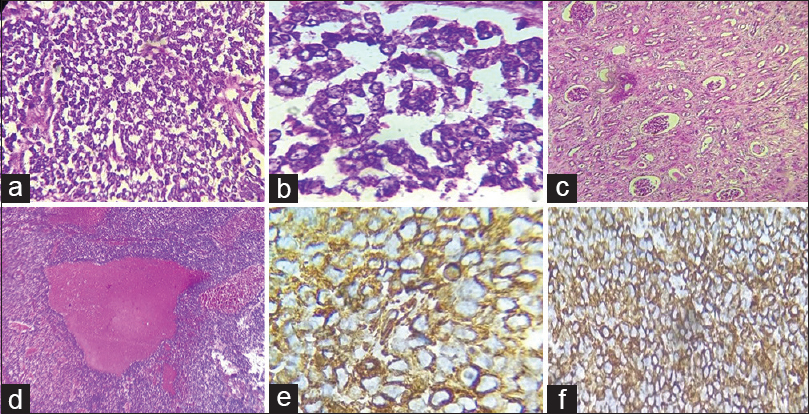
- (a) Section showing monomorphic cells with small round nuclei and fine chromatin and inconspicuous nucleoli (H and E, ×200); (b) tumor cells arranged in pseudorosettes (H and E, ×400); (c) section showing normal renal parenchyma (H and E, ×200); (d) section showing tumor with extensive hemorrhage (H and E, ×100); (e and f) figures showing positivity of tumor cells for CD99 and vimentin (e: CD99, ×400 and f: vimentin, ×200)
The patient had several episodes of cardiac asystole in the intraoperative and postoperative period and finally succumbed to the same in the intensive care unit on the 10th postoperative day.
Discussion
Primary Ewing's sarcoma (EWS)/PNET of the kidney (rPNET) is an aggressive tumor with an extremely rare occurrence. It was first described by Arthur Purdy Stout in the year 1918.[3] Since then, few case series and case reports have been published. An extensive search of the literature, <150 cases of rPNET have been reported.[12345] The largest series of rPNET in India was published by Thyavihally et al.[1] comprising 16 patients who were treated at Tata Memorial Hospital, Mumbai, Maharashtra, India. rPNET is a member of EWS family and has a neural crest cell origin. It usually occurs in childhood and in young adults, i.e., between 20 and 30 years of age and has a male: female ratio of 3:1.[123] This tumor has clinical and radiological findings like any other renal cell carcinoma. It can present with metastasis to regional lymph nodes, bone, bone marrow, lung, and liver. The study of Seth et al.[2] published a review of 382 nephrectomy specimens in a 7-year period, of which eight cases were diagnosed with rPNET. Nearly 50% (4/8) of these rPNET cases had IVC thrombi.
The patient is a 30-year-old female who presented with abdominal pain and lump of a short duration of time. Furthermore, the CECT and Doppler studies revealed a primary renal tumor, with thrombus in the IVC reaching up to the right atria.
Histologically, PNET of the kidney is composed of small-to-medium-sized monomorphic round cells with hyperchromatic nuclei and minimal cytoplasm arranged in loose cohesive sheets. Although the presence of Homer Wright rosettes is less common in extraosseous EWS, their presence is a sure/reliable diagnosis of PNET. The diagnosis is often confirmed by IHC and cytogenetics.
The reactivity to vimentin, neuron-specific enolase, and S-100 may help in making the diagnosis, but their presence is not pathognomonic. The presence of macrophage inhibitory cytokine 2 gene product, CD99 (membranous staining noted in tumor cells), confirms the diagnosis. In our case, the cells were positive for CD99/mic2, neuron-specific enolase (NSE), and vimentin and negative for cytokeratins, WT-1, synaptophysin, and chromogranin.
The major differential diagnosis of rPNET includes synovial sarcoma, adult Wilms tumor (with blastemal component only), clear-cell sarcomas, and renal neuroblastoma.[456] A panel of appropriate IHC markers as described below can help in differentiating these entities from rPNET [Table 1]. Electron microscopic findings in PNET show primitive cells, abundant cytoplasmic glycogen, and poorly developed cell junctions with no neural features.[5678]

Our case showed gross invasion of the renal pelvis and sinus by the tumor along with an IVC thrombus. Microscopy and IHC showed typical features of PNET. However, adrenals and lymph node extension/metastasis were not identified. Few case reports and short series of rPNET with IVC thrombus have been presented in the literature. Karnes et al.[7] similarly reported a case of PNET of the kidney with level II IVC thrombus. Sivaramakrishna et al.[9] reported a case of PNET of the kidney with renal vein thrombus. In their case, the patient underwent radical nephrectomy with ligation of renal vein flush on the IVC, clear of the thrombus along with chemotherapy. Seth et al.[2] reported a series of eight cases of rPNET of which four showed extension to IVC. Thyavihally et al.[1] reported rPNET with IVC thrombus in only one patient in a series of 16 patients.
Cytogenetic analysis shows EWS gene (FLI1) and ETS gene translocation t (11;22) (q24; q12). The 5-year survival of this tumor is about 45%–55%. The median relapse-free survival rate is only 2 years as reported in two case series.[1278] Most of the patients die within 1 year of diagnosis. The tumor being highly aggressive with a poor prognosis, higher rate of local recurrence, and early metastasis, surgery is the most important aspect of management followed by chemotherapy and radiotherapy.[56789] The standard chemotherapeutic agents used are as follows: vincristine (V), adriamycin (A), and cyclophosphamide (C) with or without etoposide (E) and ifosfamide (F). The patient succumbed to cardiac arrest in the early postoperative period; hence, no chemotherapy or radiation therapy could be administered to the patient.
Conclusion
PNET of the kidney is a malignant small round-cell tumor with an aggressive course of illness. PNET should be kept as a differential in mind while evaluating a large renal mass in young adults. The presence of IVC tumor thrombus portends a poor prognosis and adds to the rarity of our case. Histopathology in conjunction with IHC helps in correctly labeling this entity, thereby guiding the clinicians to take early and aggressive therapeutic intervention.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Primitive neuroectodermal tumor of the kidney: A single institute series of 16 patients. Urology. 2008;71:292-6.
- [Google Scholar]
- Primitive neuroectodermal tumors of kidney: Our experience in a tertiary care center. Indian J Cancer. 2016;53:109-12.
- [Google Scholar]
- Primary Ewing's sarcoma/primitive neuroectodermal tumor of the kidney: A clinicopathologic and immunohistochemical analysis of 11 cases. Am J Surg Pathol. 2002;26:320-7.
- [Google Scholar]
- Primitive neuroectodermal tumor (PNET) of kidney with level IV inferior venacaval thrombus: A case report. Recent Res Sci Technol. 2010;2:38-41.
- [Google Scholar]
- Primitive neuroectodermal tumor (Extraskeletal Ewing's sarcoma) of the kidney with vena caval tumor thrombus. J Urol. 2000;164:772.
- [Google Scholar]
- Primary primitive neuroectodermal tumor of the urinary tract. J Formos Med Assoc. 2006;105:1008-12.
- [Google Scholar]
- Primary primitive neuroectodermal tumor (PNET) of the kidney with venous thrombus. Int Urol Nephrol. 2003;35:311-2.
- [Google Scholar]



