
Translate this page into:
Transfusion-acquired Hemoglobinopathies: A Report of Two Cases
Address for correspondence: Dr. Abhishek Purohit, E-mail: purohitabhi80@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Transfusion-acquired hemoglobinopathy occurs when a carrier of hemoglobinopathy with no significant abnormalities donates blood, and the blood is transfused to a recipient. This process can lead to spurious results in the recipient without any clinical abnormality or infrequently can result in disastrous situations. The incidental finding of such posttransfusion related abnormal peaks in hemoglobin high-performance liquid chromatography (Hb HPLC) may cause diagnostic dilemmas and result in unnecessary laboratory testing. Here, we report two such cases of transfusion-acquired hemoglobinopathies, which were subsequently resolved by the abnormally low percentage of the Hb variants, transient nature of the peaks, and parental Hb HPLC.
Keywords
High-performance liquid chromatography
posttransfusion peak
transfusion-acquired hemoglobinopathy

INTRODUCTION
Transfusion-acquired or apparent hemoglobinopathy is an increasingly recognized entity with the widespread availability of high-performance liquid chromatography (HPLC). Asymptomatic carriers of abnormal hemoglobin (Hb) may donate blood, which may not be detected by the routine blood donor screening process. Such blood, when transfused, may cause aberrant results in the recipients. There are few case reports and very few case series available in this regard.[123456]
Here, we report two cases of β-thalassemia major, which revealed abnormal transfusion-associated peaks during work up.
CASE REPORT
First case, an 8-year-old male child was brought with history of transfusion-dependent anemia since 6 months of age with an hemoglobin (Hb) of 8.4 g/dL and peripheral smear showing microcytic hypochromic red cell picture [Table 1]. Considering the possibility of thalassemia major and unavailability of prior Hb HPLC report, Hb HPLC (BioRad Variant II, β- thalassemia Short Program) was carried out in our department, which showed a HbF level of 1.9%, with HbA0 73.0%, HbA2 2.4% and a peak in D-window of 8.9% with a retention time of 4.07 min [Figure 1a]. Percentage of HbD in heterozygotes for HbD approximates 40%, hence, a possibility of transfusion-acquired HbD was considered. Hb HPLC of both parents revealed heterozygous state for β-thalassemia [Figure 1b and c]. However, there was no abnormal peak at D-window in both the parents. A repeat Hb HPLC on patient's blood was carried just prior to the next transfusion, which revealed a decline in the percentage of the peak in D-window to 1.6%. The patient was on regular transfusion and had received transfusion 2 weeks prior to the first Hb HPLC, which could have been the reason for the transfusion-associated peak.
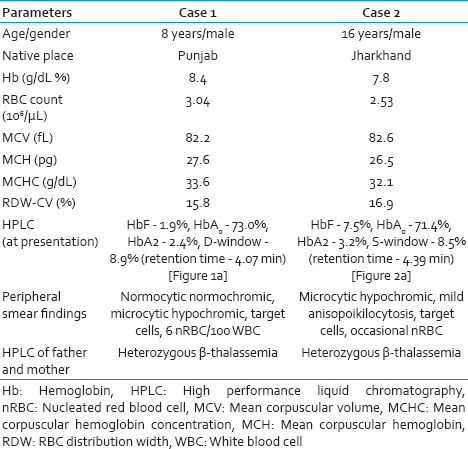
- (a) Hemoglobin high-performance liquid chromatography of patient showing a peak in D-window of 8.9% with a retention time of 4.07 min. Chromatogram of father (b) and mother (c)
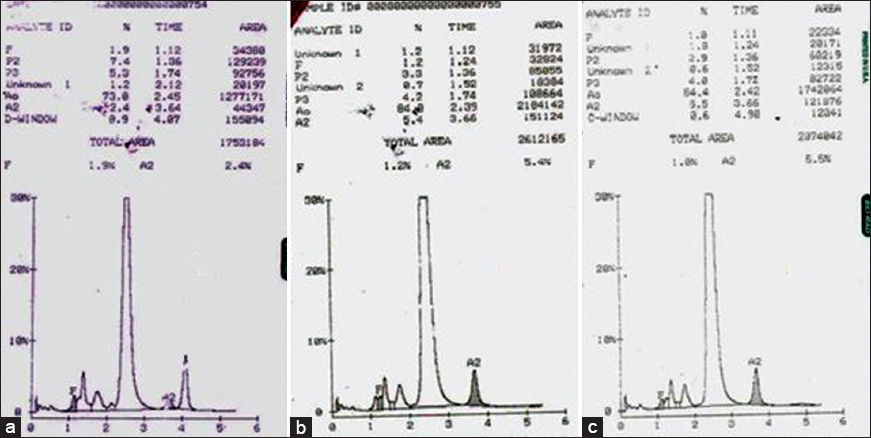
Similarly, the second case was a 16-year-old boy, who presented with a history of transfusion-dependent anemia since 1-year of age. His hemogram revealed a Hb of 7.8 g/dl and peripheral smear showed anisopoikilocytosis and microcytic hypochromic red cell picture [Table 1]. Hb HPLC of the patient showed raised HbF level of 7.5% with HbA0 71.4%, HbA2 3.2% and a peak in S-window of 8.5% with a retention time of 4.39 min [Figure 2a]. The expected percentage of HbS in heterozygotes is approximately 40% unless there is an associated α gene deletion. Hb HPLC of both parents showed heterozygous state for β-thalassemia [Figure 2b and c]. However, there was no abnormal peak at S-window in both the parents. As the patient had received transfusion 10 days prior to Hb HPLC, a repeat Hb HPLC on patient's blood was carried out after a month, which showed fall in peak in S-window to 1.0% indicating the transient nature of this transfusion-associated peak. During this intervening 1-month, the patient had not received a transfusion.
- (a) Hemoglobin high-performance liquid chromatography of patient showing a peak in S-window of 8.5% with a retention time of 4.39 min. Chromatogram of father (b) and mother (c)
DISCUSSION
Transfusion-acquired hemoglobinopathy is not uncommon and frequently lead to misdiagnosis, repeated testing, extensive counseling of patients. HPLC as a method of identification and quantification of Hb variants has improved precision and accuracy when compared with the established method of hemoglobin electrophoresis. With the widespread availability of Hb HPLC, transfusion-acquired hemoglobinopathies caused by blood transfusions are becoming far more common than previously reported.[1]
Kozarski et al. reported one of the largest series of apparent hemoglobinopathies with 52 occurrences of transfusion-associated peaks in 32 recipients with HbC being the most common transfused Hb detected in their study.[1] Suarez et al. had reported a case of blood transfusion-acquired HbC in which the observed concentration of HbC was 10.9%.[2] Another incidence of acquired HbC was reported by Strobel et al. in a 52-year-old male and the suspicion, in this case, was raised by an abnormal non-lysis of RBCs detected by automated cell counter.[3]
Gupta et al. in their study, of 1530 Hb HPLC over 1½ years identified three cases of posttransfusion hemoglobinopathy with all cases showing HbS in the range of 9.9–18.5%.[4] Jain et al. reported one case of posttransfusion peak in a case of juvenile myelomonocytic leukemia with HbD of 10.2%.[5] Chauhan et al. had reported a case of posttransfusion peak of 9.1% Hb Q India with a retention time of 4.71 min.[6]
It is important to make the diagnosis of a hemoglobinopathy, as well as to avoid misdiagnosis, of a hemoglobinopathy as brought out by Wong et al. when a diagnosis of heterozygous β-thalassemia was made in an otherwise normal individual after receiving transfusion from HbE carrier.[7] Rarely, transfusion of abnormal Hbs can lead to catastrophe as in a case of a neonate transfused with blood from a sickle cell trait donor, which resulted in multiple renal and splenic infarcts in the recipient.[8]
In our report of two cases, the percentages of abnormal hemoglobin were 8.9% of HbD and 8.5% of HbS, respectively. The possibility of transfusion-associated peaks in these cases was suspected and proved on the basis of low concentrations of abnormal Hbs, transient nature of the peaks whose concentrations have reduced on subsequent testing and the absence of similar peaks in their parents. The almost similar strategy was used in the above-mentioned studies though rarely investigators had resolved to molecular techniques to resolve the issue, which can result in unnecessary testing, improper counseling, wastage of time, and resources. Hence, a careful history regarding blood transfusion, avoidance of prescribing Hb HPLC in a recently transfused individual and in unavoidable situations performing the chromatography along with parental study can prevent such diagnostic dilemmas.
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- Blood transfusions leading to apparent hemoglobin C, S, and O-Arab hemoglobinopathies. Arch Pathol Lab Med. 2006;130:1830-3.
- [Google Scholar]
- Hemoglobin C acquired via a blood transfusion. Detection by automated blood counter. Arch Pathol Lab Med. 1987;111:565-8.
- [Google Scholar]
- Transfusion-induced hemoglobinopathy in patients of beta-thalassemia major. Indian J Pathol Microbiol. 2011;54:609-11.
- [Google Scholar]
- Transfusion associated peak in hb HPLC chromatogram – A case report. Mediterr J Hematol Infect Dis. 2012;4:e2012006.
- [Google Scholar]
- Transfusion associated unknown peak on HPLC chromatogram: A diagnostic dilemma. J Appl Hematol. 2014;5:19-22.
- [Google Scholar]
- A blood transfusion leading to misdiagnosis of beta-thalassaemia carrier status. Blood Transfus. 2010;8:69-70.
- [Google Scholar]
- Multiple renal and splenic infarctions in a neonate following transfusion with sickle trait blood. Clin Pediatr (Phila). 1982;21:239-41.
- [Google Scholar]



