
Translate this page into:
Acquired aplastic anemia associated with trisomy eight converting into acute myeloid leukemia
Address for correspondence: Dr. Sumit Grover, Department of Pathology, Dayanand Medical College and Hospital, Ludhiana, Punjab, India. E-mail: sumitgrover1204@gmail.com
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Aplastic anemia (AA) is nowadays considered to be a clonal disorder arising from a defective hematopoietic stem cell developing after a generalized insult to bone marrow. Immunosuppressive treatment (IST) of AA causes suppression of the target dominant population of haematopoietic cells allowing the defective non targeted clones to expand. This may give rise to acute leukemia. Cytogenetic studies for chromosomal aberrations such as trisomy and monosomy may help in detecting such conversions. We present a case of acquired AA in a 60-year-old male presenting with pancytopenia and hypoplastic marrow treated with antithymocyte globulin, converting into myelodysplastic syndrome and later on acute promyelocytic leukemia after being in remission for 4 years. The patient was found to have trisomy 8 on fluorescence in situ hybridization and karyotyping.
Keywords
Acute myeloid leukemia
aplastic anemia
clonal
trisomy 8

Introduction
A plastic anemia (AA) is defined by pancytopenia and hypoplastic bone marrow which may result from a generalized insult to bone marrow. AA may run an acute course, a subacute course, or a chronic course with episodes of transient remission in-between. However, in rare cases, a conversion to other related hematological disorders such as myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML) is seen.[12] This happens due to abnormal clonal proliferation induced by IST within a span of 6 months to 20 years.[3] We present a rare case in which AA transformed into MDS followed by AML with in a duration of 4 years.
Case Report
Case 1
A 60-year-old male presented with progressive breathlessness and petechial spots in 2010. The investigations showed hemoglobin of 5.0 g/dl, white blood cell count of 2.6 × 109/l, platelet count of 3 × 109/l, and reticulocyte count of 0.1%. The bone marrow aspirate (BMA) smears showed markedly hypoplastic fragments with diminished hematopoiesis and absence of atypical cells [Figure 1a]. The bone marrow trephine biopsy revealed markedly reduced cellularity with mostly fat spaces and occasional lymphoid aggregate confirming AA [Figure 1b]. Flow cytometry showed CD55, CD58, CD59, and Gly A on 99% of red blood cells ruling out the possibility of paroxysmal nocturnal hemoglobinuria (PNH). The patient was treated with antithymocyte globulin (ATG), cyclosporine, and prednisolone but no colony stimulating factors (CSFs). After 4 years of remission, he developed herpes zoster and sinonasal zygomycosis. The peripheral blood counts revealed again pancytopenia but with 10% blast cells and a few abnormal promyelocytes. BMA showed a hypercellular marrow with presence of abnormal promyelocytes and 12% blast cells. A diagnosis of MDS-refractory anemia with excess of blasts was kept. Cytogenetic studies (fluorescence in situ hybridization [FISH]) and karyotyping showed trisomy 8. However, FISH was negative for monosomy 5, monosomy 7, or deletion 5q, 7q, and 20q. Three months later, the patient was again admitted to the emergency with petechial spots, altered sensorium. Peripheral blood showed 27% blasts and 33% abnormal promyelocytes confirming the conversion of MDS to acute leukemia. BMAs and biopsy were hypercellular and packed with atypical promyelocytes and blasts [Figure 1c–f]. Flow cytometry showed HLADR, CD117, CD33, CD13, and myeloperoxidase positivity in gated cells of the blast region confirming myeloid lineage [Figure 2].
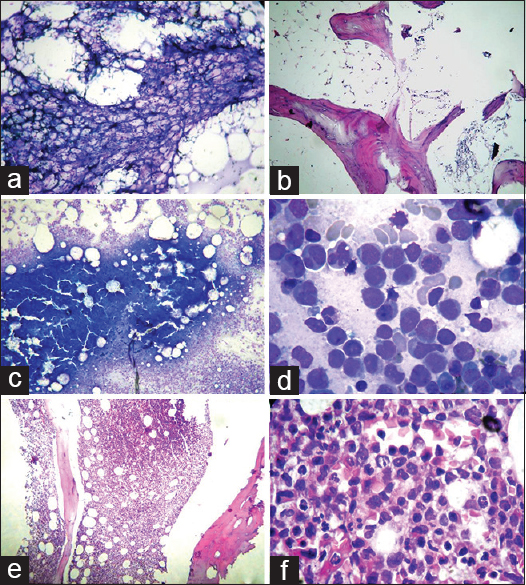
- (a and b) Hypocellular bone marrow aspirate (Giemsa, ×100) and biopsy (H and E, ×100), (c) hypercellular marrow aspirate (Giemsa, ×100), (d) atypical promyelocytes and blasts (Giemsa, ×1000), (e) hypercellular bone marrow biopsy (H and E × 100), (f) atypical promyelocytes on biopsy (H and E, ×1000)
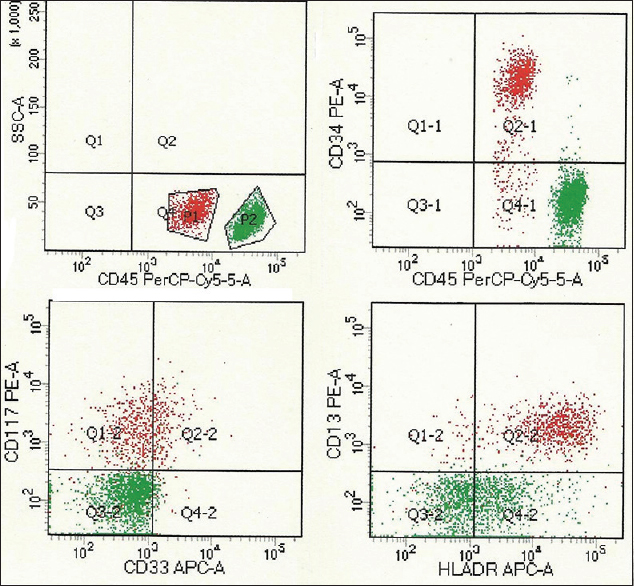
- CD45 dim red cluster depicts myeloid blasts with positivity for CD34, HLADR, CD13, CD33, and CD117
Discussion
AA is closely related to and shares common pathophysiologic features with other bone marrow failure syndromes such as MDS and PNH. All three diseases may present with pancytopenia and a hypocellular bone marrow. According to Brodsky, AA, MDS and leukaemia represent different manifestations of generalized insults to the bone marrow. A primary insult to the bone marrow could simultaneously lead to several abnormal hematopoietic cell clones, one dominant and the other present below the level of detection. Immunosuppressive therapy in AA suppresses the dominant clone allowing the abnormal clones to expand and becomes detectable.[4] Hypoplastic MDS may be difficult to distinguish from AA based on morphology alone. Megakaryocytes are the most reliable lineage to use in distinguishing MDS from severe AA; small mononuclear or aberrant megakaryocytes are typical of MDS, whereas megakaryocytes are markedly reduced or absent in severe AA.[5] Cytogenetics are helpful when typical of MDS, but some aberrations (such as trisomy 6, trisomy 8, and 13q−) may appear in severe AA that is responsive to IST.[6] However detection of cytogenetic abnormalities in severe AA may be difficult because of difficulties in obtaining sufficient cells for analyses.[7] In our case too initially karyotyping had uninformative results. But patient achieving complete remission for four years on IST supported diagnosis of AA. Trisomy 8 was detected when patient relapsed with MDS (RAEB).
Clonal evolution occurs in approximately 10% to 15% of patients of severe AA and usually manifests as worsening blood counts unresponsive to immunosuppression, prominent dysplastic findings in the bone marrow, and abnormal cytogenetics.[5] Karyotypes most frequently encountered in MDS and AML secondary to AA involve chromosomes 6, 7 and 8. Xiao Y et al and Ma Y et al concluded trisomy 8 as the most frequent cytogenetic abnormality with a prevalence of 14.21% (n = 168) and 16.7% (n = 435) respectively. They considered it an intermediate cytogenetic risk based on International Prognostic Scoring System (IPSS) while assessing prognostic role of +8 in MDS patients by comparing patients with normal karyotype, 20q- and -7/7q-[89]
Hypoplastic MDS and AA have different risk of developing AML (83% vs. 9%).[10] Wang H et al found 9.2% patients of primary MDS progressing to AML within 4.5 years and found trisomy 8 to be a common association.[11] Li Y et al and Ogha et al found incidence of 1.7% and 22% at 5 years for clonal evolutions to MDS/AML in 802 and 50 AA patients respectively after treatment with cyclosporin and G-CSF.[1213] IST including ATG and cyclosporine have increased the risk of late clonal haematological complications.[14] Li Y et al concluded number of days of G-CSF therapy as risk factor for clonal evolution.[12] Whether this is treatment related or due to longer survival after IST leading to growth of the abnormal clone of defective stem cells is not clear. Our case showed conversion to AML after a period of 4 years of complete remission on IST but without administration of G-CSF. Thus trisomy 8 in our case was associated with MDS/AML secondary to AA.
Conclusion
As long-term use of cytokines and IST in patients with severe aplastic anaemia may induce or hasten the onset of a malignant transformation, careful attention must be paid to clonal evolution. Due to the poor prognosis of secondary myelodysplasia and leukaemia, allogenic bone marrow transplantation for such patients must be carried out early in the course of the disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Evolution of clonal cytogenetic abnormalities in aplastic anemia. Leuk Lymphoma. 2004;45:433-40.
- [Google Scholar]
- Aplastic anaemia evolving into overt myelodysplastic syndrome/acute myeloid leukaemia with t(3;5)(p25;q31) Cancer Genet Cytogenet. 2002;137:91-4.
- [Google Scholar]
- The problem of clonality in aplastic anaemia: Dr Dameshek's riddle, Restated. Blood. 1992;79:1385-92.
- [Google Scholar]
- Riddle what do aplastic anaemia and acute promyelocytic leukaemia, and chronic myeloid leukaemia have in common. Leukaemia. 2004;18:1740-2.
- [Google Scholar]
- Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92-6.
- [Google Scholar]
- Cytogenetic profile of aplastic anaemia in Indian children. Indian J Med Res. 2013;137:502-6.
- [Google Scholar]
- Trisomy 8 is the most frequent cytogenetic abnormality in de novo myelodysplastic syndrome in China. Onkologie. 2012;35:100.
- [Google Scholar]
- Prognostic value of trisomy 8 in primary myelodysplastic syndrome. Intern Med J. 2010;40:697-703.
- [Google Scholar]
- Myelodysplastic syndrome and aplastic anaemia: Distinct entities or diseases linked by a common pathophysiology? Semin Hematol. 2000;37:15-29.
- [Google Scholar]
- Cytogenetic features and prognosis analysis in Chinese patients with myelodysplastic syndrome: A multicenter study. Ann Hematol. 2010;89:535-44.
- [Google Scholar]
- Long-term follow-up of clonal evolutions in 802 aplastic anaemia patients: A single-center experience. Ann Hematol. 2011;90:529-37.
- [Google Scholar]
- Treatment responses of childhood aplastic anaemia with chromosomal aberrations at diagnosis. Br J Haematol. 2002;118:313-9.
- [Google Scholar]
- Risk factors for evolution of acquired aplastic anaemia into myelodysplastic syndrome and acute myeloid leukaemia after immunosuppressive therapy in children. Blood. 2002;100:786-90.
- [Google Scholar]



