
Translate this page into:
Cytopathological examination of bronchoalveolar lavage fluid in diagnosis of pulmonary alveolar proteinosis
Address for correspondence: Dr. Manjari Kishore, A1, 1/10A, Sector-5, Rajender Nagar, Sahibabad, Ghaziabad, Uttar Pradesh, India. E-mail: drmanjarik@gmail.com
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Pulmonary alveolar proteinosis (PAP) is a rare disease characterized by the deposition of extracellular lipoproteinaceous material within the air spaces. Although the diagnosis is mainly based on histopathological findings, sometimes, the diagnostic yield of transbronchial and even open lung biopsy can be unsatisfactory. The advantage with bronchoalveolar lavage (BAL) cytology is that apart from being safer for the patient, it can sample a much wider area and help in giving an early diagnosis and treatment to the patient. Herein, we present a case of PAP diagnosed on BAL fluid cytology in an elderly female.
Keywords
Alveolar proteinosis
bronchoalveolar lavage
cytomorphology
lung

Introduction
Pulmonary alveolar proteinosis (PAP) is a rare disease with no exact etiology known.[1] Multiple factors are involved in its pathogenesis resulting in progressive filling of the pulmonary alveoli with amorphous, periodic acid–Schiff (PAS)-positive diastase-resistant, lipoproteinaceous, surfactant-related material.[12] Varied clinical presentation is seen in the patients of PAP such as progressive dyspnea on exertion, chronic nonproductive cough, fatigue, mild weight loss, chest pain, and low-grade fever. Bronchoalveolar lavage (BAL) fluid examination is a noninvasive procedure. It helps in the diagnosis of PAP by sampling a wider area, especially in cases where biopsy shows inconclusive result. We present a case of PAP diagnosed based on the cytomorphological diagnosis of BAL fluid in an elderly female with underlying interstitial lung disease where biopsy was inadequate.
Case Report
A 52-year-old female presented to the hospital with complaints of on and off breathlessness and dry cough for the past 3–4 months along with fever for the past 1–2 days. She had a similar history of breathlessness 2–3 years back. However, there was no any history of hemoptysis, joint pain, rashes, smoking, or any other chronic illness. General examination of the patient did not reveal any abnormality. However, examination of respiratory system revealed rhonchi on intrascapular and bilateral supramammary regions (more on the left side). Two-dimensional echocardiography showed normal cardiac chamber dimensions along with normal cardiac valves. Rest of the systems examined were within normal limit. Her saturation levels were 99% (on oxygen) and 84% (off oxygen).
Laboratory investigations such as complete blood count, random blood sugar, and renal and liver function tests were within normal range as defined for her age. No abnormality was noted on fiber-optic bronchoscopic examination. However, X-ray chest done revealed patchy opacity on the left midzone and right paracardiac region with normal cardiac shadows. An impression of pneumonitis and alveolar proteinosis was given based on X-ray findings [Figure 1a]. These findings were further confirmed on contrast-enhanced computed tomography (CECT) chest which showed bilateral ground-glass haze in the lung parenchyma with interlobular septal thickening. The characteristic crazy pavement was noted on CECT chest [Figure 1b and c]. A possibility of alveolar proteinosis was given on CECT, and a correlation with BAL was advised.
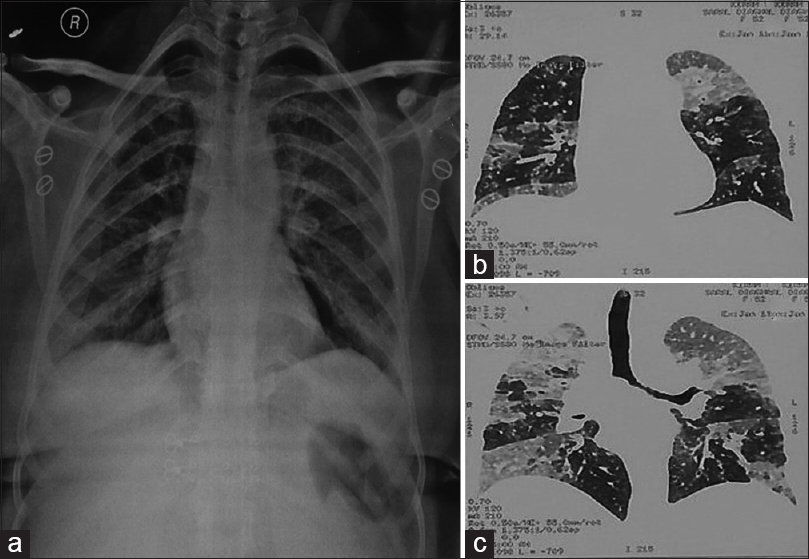
- (a) X-ray chest showing patchy opacity on the left midzone and right paracardiac region (pneumonitis) along with left hilar and right basal infection (alveolar proteinosis). (b and c) contrast-enhanced computed tomography chest showing bilateral ground-glass haze in lung parenchyma with interlobular septal thickening along with characteristic crazy pavement was noted on contrast-enhanced computed tomography chest suggesting a possibility of alveolar proteinosis
BAL was performed and 4 ml of thick proteinaceous fluid was received. Gross examination of fluid was also consistent with fluid as seen in PAP. Smears examined showed plaques of eosinophilic material and few groups of macrophages filled with the similar eosinophilic material [Figure 2a-c]. These plaques were positive for PAS [Figure 2d]. Smears also revealed acute and chronic inflammatory cells with marked eosinophilia along with few singly scattered respiratory epithelial cells [Figure 2a, inset]. Based on the clinical, radiological, and cytomorphological findings, a diagnosis of PAP was given. The patient was given symptomatic treatment with oxygen inhalation @ 2 L/min and antibiotic therapy. The patient is kept under follow-up and currently doing well with conservative treatment.
![(a and b) Photomicrograph showing plaques of eosinophilic material and few groups of macrophages filled with the similar eosinophilic material. (a and b) Papanicolaou ×20 and ×40). Inset ([a] Few benign endobronchial cells). (c) Smear revealing few plaques of eosinophilic material and background showing inflammatory cells (Giemsa, ×40). (d) Eosinophilic globules showing periodic acid–Schiff positivity (×40)](/content/164/2018/10/1/img/JLP-10-109-g002.png)
- (a and b) Photomicrograph showing plaques of eosinophilic material and few groups of macrophages filled with the similar eosinophilic material. (a and b) Papanicolaou ×20 and ×40). Inset ([a] Few benign endobronchial cells). (c) Smear revealing few plaques of eosinophilic material and background showing inflammatory cells (Giemsa, ×40). (d) Eosinophilic globules showing periodic acid–Schiff positivity (×40)
Discussion
PAP is a rare disorder described for the first time in 1958 in a patient with no any underlying abnormality.[1] This entity is characterized by deposition of a granular extracellular material composed of PAS positive, proteins and lipids, and diastase resistant. No exact etiology is known.[2] However, these excessive intra-alveolar accumulations of phospholipids in PAP are due to increased production and secretion of surfactant phospholipids by Type II pneumocytes.[3] Other factors which are implicated in its pathogenesis are defective clearance of alveolar surfactant and defective alveolar macrophage function.[3]
The usual age group affected by PAP is between 20 and 50 years but can be noted in neonates and even elderly. Men are affected more than women (M:F: 2–4:1).[3] PAP may be either idiopathic or sometimes associated with immunosuppression or chronic exposure to organic dusts such as silica, aluminum dusts, or various pulmonary infections such as Nocardia, Pneumocystis carinii, cytomegalovirus, tuberculosis, and Histoplasma capsulatum.[4]
These findings can be confirmed by the transbronchial lung biopsy or open lung biopsy specimen. However, problem arises when transbronchial biopsy is very small and cannot provide exact evidence. It is also important to note that the characteristic intra-alveolar material is localized mainly in the pulmonary hilar region, so sometimes even open lung biopsy may not show conclusive results. In the present case, also, transbronchial biopsy did not show any definitive result. The advantage with BAL fluid is that apart from being noninvasive, much larger area can be sampled. Although the definitive diagnosis is based on the ultrastructural findings, it is not practical to process all such specimens for electron microscopy. However, the presence of the characteristic multilamellated structure under the electron microscope provides the confirmative diagnosis. Mikami et al. had reported three cases of PAP diagnosed on BAL fluid cytology along with detailed description of ultrastructural findings seen in alveolar proteinosis.[4]
Maygarden et al. reported complete cytological, histochemical, and ultrastructural findings in BAL fluid.[5] Takemura et al. had examined the ultrastructural findings from the sediments of BAL fluid and categorized the multilamellated structures into four types as Type A-D:[6]
-
Type A: Major component consisted of concentric trilaminar structures which comprised two electron-dense layers and a central lucent layer (5.7–7.5 nm) alternating with wider (25–30 nm) electron-lucent layers
-
Type B: Formed by concentric lamellae with 5.0–5.3 nm periodicity
-
Type C: Composed of wavy, electron-dense lamellae with a 4.0–4.5 nm periodicity
-
Type D: Conglomerated masses of intricately arranged double or triple electron-dense layers (7.5–13.5 nm wide) alternating with wider (30–40 nm) electron-lucent layers.
As a part of investigation and management, BAL was sent for the examination. Grossly, BAL fluid was opaque which is consistent with fluid as seen in PAP. This simple procedure serves as a mode of treatment in the PAP case, especially in debilitated/transplant patients.
In the present case, PAS-positive globules confirmed the diagnosis of PAP. However, before giving a definitive diagnosis, it is important to keep in mind the other differentials which can mimic the globules as seen in PAP. The first entity which must be kept in mind is Pneumocystis carinii pneumonia (PCP), and it shows foamy mass/exudate with distinct dark dots in individual vacuoles even in Papanicolaou smears. However, these globules can be differentiated from PAP globules using PAS and Grocott's methenamine silver (GMS) stains. The globules of PCP will show GMS-positive organism with crushed ping-pong ball-like appearance in the frothy cast. Kotov et al. had reported a case of alveolar proteinosis in a patient recovering from Pneumocystis carinii infection and concluded that a relationship exists among the two diseases, especially in an immunocompromised patient.[1] Hence, an utmost care should be taken to distinguish these globules in both the entities using special stains as mentioned above and correctly labeling them.
Although amyloidosis would be an unusual finding in BAL fluid, sometimes, the globules of PAP should be differentiated from amyloid deposits which show amorphous acellular material which may cytologically[7] resemble extracellular material as noted in PAP. However, these globules are PAS negative and show Congo red positivity with orange-red stain along with typical apple-green birefringence under polarized light. The ultrastructural findings of amyloidosis are also helpful showing interlacing meshwork of nonbranching fibrils.
Other differentials that should be kept in mind are resolving pneumonia, drug-related pulmonary toxicity (amiodarone and chlorpheniramine).[8] A proper clinical history and important cytological findings such as vacuolated histiocytes in drug-related toxicity can help in distinguishing them from PAP.[8]
The removal of accumulated intra-alveolar lipoproteinaceous material through bronchoscopic BAL is an effective mode of treatment, as done in the present case. This leads to improved pulmonary function.[89] As clinical course of PAP is highly variable, sometimes, the patient may present with severe hypoxemia, cor pulmonale, or even respiratory failure in approximately 15%–20% of the patients. Very rarely, death may occur due to superimposed infections.[89] Our patient responded well to the conservative management and is still under follow-up and presently doing well.
Conclusion
PAP is a potentially fatal though rare lung disorder and radiographically may mimic other common types of pulmonary disease. The examination of BAL fluid may result in an accurate and prompt diagnosis of PAP, eliminating the need of much invasive procedures. Hence, an early and effective management can be given to these patients, thereby decreasing the morbidity and mortality.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Alveolar proteinosis in a patient recovering from pneumocystis carinii infection: A case report with a review of literature. Cytojournal. 2006;3:22.
- [Google Scholar]
- Cytohistologic and electron microscopic findings in bronchoalveolar lavage fluid in a case of pulmonary alveolar proteinosis. Diagn Cytopathol. 2002;27:63-5.
- [Google Scholar]
- Pulmonary alveolar proteinosis: Diagnosis using routinely processed smears of bronchoalveolar lavage fluid. J Clin Pathol. 1997;50:981-4.
- [Google Scholar]
- Pulmonary alveolar proteinosis: A spectrum of cytologic, histochemical, and ultrastructural findings in bronchoalveolar lavage fluid. Diagn Cytopathol. 2001;24:389-95.
- [Google Scholar]
- Ultrastructural, histochemical, and freeze-fracture evaluation of multilamellated structures in human pulmonary alveolar proteinosis. Am J Anat. 1987;179:258-68.
- [Google Scholar]
- Autoantibodies against granulocyte macrophage colony-stimulating factor are diagnostic for pulmonary alveolar proteinosis. Am J Respir Cell Mol Biol. 2002;27:481-6.
- [Google Scholar]
- Cytopathology of pulmonary alveolar proteinosis complicating lung transplantation. J Heart Lung Transplant. 2004;23:135-8.
- [Google Scholar]



