
Translate this page into:
Development of a dry-reagent mix-based polymerase chain reaction as a novel tool for the identification of Acinetobacter species and its comparison with conventional polymerase chain reaction
Address for correspondence: Dr. Shama Bhat, Bhat Biotech India Pvt. Ltd., 11-A, 4th Cross, Veerasandra Industrial Area, Electronic City Phase II, Bengaluru - 560 100, Karnataka, India. E-mail: bhatbiotech@gmail.com
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
BACKGROUND:
Nosocomial infections are often caused by multidrug-resistant bacteria and the incidence is increasing. Acinetobacter, a Gram-negative bacillus, is commonly associated with the use of intravascular catheterization and airway intubation. Polymerase chain reaction (PCR) for identification of Acinetobacter baumannii from samples has been standardized that use conventional wet-reagent mix. We have designed and optimized a dry-reagent mix for identification of Acinetobacter species by PCR. The dry-reagent mix can be stored at room temperature, has less chances of contamination, and thus can be used at point-of-care diagnosis.
AIM AND OBJECTIVE:
The present work was focused on comparing the sensitivity and specificity of dry-reagent PCR mix over conventional wet-reagent PCR mix for identification of Acinetobacter species.
MATERIALS AND METHODS:
Conventional wet-reagent mix based and dry-reagent mix based PCR were carried out for the DNA isolated from Acinetobacter species. The latter was also applied directly on bacterial growth without prior DNA extraction process. Equal numbers of bacterial isolates other than Acinetobacter species were also subjected to identification by the same protocols for determining the sensitivity and specificity of the test.
RESULTS:
The Acinetobacter species showed amplification of the target rpoB gene and the band was observed at 397 bp. The dry-reagent PCR mix results matched completely with the conventional wet-reagent PCR mix assay. All the non-Acinetobacter isolates were negative for the PCR. This indicates that the test is highly specific. The dry-reagent mix also contained an enzyme resistant to PCR inhibitors and capable of amplifying DNA directly from cells.
CONCLUSION:
Performance of dry-reagent PCR mix without the need for DNA extraction and preparation of a PCR mix proved to be more sensitive and reduce the handling error, minimizes the time, manual work, and skilled labor.
Keywords
Acinetobacter
DNA
dry-reagent mix
inhibitors
polymerase chain reaction

Introduction
Acinetobacter, a Gram-negative coccobacillus, is emerging as an important cause of nosocomial infections and is one of the highly drug resistant organisms.[123] In most clinical laboratories, the identification of Acinetobacter species is generally done by phenotypic methods. Molecular identification of Acinetobacter amplifying the rpoB gene has been well evaluated and found to be highly specific.[4] The reaction targets a 350 bp amplicon size of rpoB gene.[145] Although polymerase chain reaction (PCR) is a highly reliable and most sensitive technique and helps in the rapid identification of bacteria, it requires extraction of DNA from the bacterial isolates/clinical samples and use of freshly prepared reagents. Whereas, in the case of the dry-reagent mix, it is reconstituted by adding DNase-free water just few minutes before the PCR is run.[6] This minimizes the contamination and pipetting errors. There are several earlier literature available on freeze-dried reagent mix for PCR-based diagnosis of infectious diseases such as HIV-1, HCV, CMV, and Mycobacterium tuberculosis. They state that the use of dry-reagent mix is cost-effective, time efficient, stable at room temperature, less demanding, and easy to handle, thus useful in low-resource settings.[7891011]
We report here successful use of dry-reagent mix-based PCR for identification of Acinetobacter species. In the present work, we have compared the performance of our proprietary dry-reagent mix against conventional wet-reagent mix for the identification of Acinetobacter species. We have compared with both DNA as well as direct bacterial culture without prior DNA extraction using the dry-reagent mix.
Materials and Methods
The study was conducted at SDM College of Medical Sciences and Hospital, Dharwad, from March 2016 to April 2017. A total of 200 isolates, 100 phenotypically confirmed Acinetobacter isolates and 100 phenotypically confirmed non-Acinetobacter bacterial isolates (Citrobacter diversus – three, Citrobacter freundii – three, coagulase-negative staphylococci– four, Escherichia coli – eight, Enterococcus species – five, Enterobacter cloacae – one, Enterobacter species – two, Enterococcus faecalis – five, Klebsiella pneumonia – ten, methicillin-resistant Staphylococcus aureus – five, nonfermenting Gram-negative bacilli – seven, Proteus mirabilis – four, Proteus vulgaris – four, Pseudomonas aeruginosa – eight, Pseudomonas species – three, Salmonella spp. – four, Salmonella typhi – three, Shigella flexneri – four, S. aureus – ten, Streptococcus species – seven, and Vibrio cholerae – two), from various clinical samples were included in the study. All these isolates were identified to genus level by conventional methods such as colony morphology, Gram-staining, catalase, oxidase, and motility.
Bacterial genomic DNA isolation
A single colony from pure subcultures was inoculated in one ml Luria Bertani (LB) broth in sterile microcentrifuge tube and incubated overnight. The tubes were centrifuged at 10,000 rpm for ten min, and the supernatant was discarded to harvest bacterial pellet. The bacterial pellet was resuspended in 300 μl of suspended in extraction buffer (75 mM NaCl; 25 mM Na2 ethylenediaminetetraacetic acid; pH 8.0) in an Eppendorf tube. To each tube, 15 μl of 1% sodium dodecyl sulfate and 20 μl of proteinase K were added. The tubes were then incubated at 50°C for 2 h. To the lysate, 400 μl of phenol-chloroform (1:1) was added and the tube was vortexed. The lysate mixture was centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was carefully transferred to a fresh Eppendorf tube and equal volume of chloroform-isoamyl alcohol (24:1) was added and the tube was vortexed and centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was carefully transferred to fresh Eppendorf tubes, and 0.1 volume of 3M sodium acetate and two volume of prechilled absolute alcohol were added. The contents in the tubes were mixed gently and incubated at −20°C for 2 h. After incubation, it was centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was discarded and the pellet was washed with one ml of 70% ice-cold ethanol, vortexed and centrifuged at 13,000 rpm for three min at 4°C. The supernatant was discarded and the pellet was dried at 40°C, with the lids of the tubes open. The pellet was resuspended in 100 μl of sterile PCR grade water and incubated for 1 h at 37°C.[12] The purified DNA was used as template for both conventional wet-reagent PCR mix and dry-reagent PCR mix assays.
Primers and conventional polymerase chain reaction conditions
Following primers were used in the PCR assays, namely, forward primer 5’ TAYCGYAAAGAYTTGAAAGAAG 3’ and reverse primer 5’ CMACACCYTTGTTMCCRTGA 3’[5] which targets 397 bp region of the rpoB gene of Acinetobacter. The DNA isolated from all the 200 bacterial isolates was subjected to both conventional wet-reagent mix and dry-reagent PCR mix while bacterial cultures in LB broth were tested by dry-reagent PCR mix. A. baumannii (ATCC) was used as reference control in all experiments. Conventional wet-reagent mix consisting of 1X reaction buffer (Fermentas), 2 mM MgCl2 (Fermentas), 0.5 mM each dNTP (Fermentas), 0.5 μM each of forward and reverse primer (Sigma-Aldrich), 1 U of Taq polymerase (Fermentas) was prepared and 2 μl of isolated template DNA was added and the reaction volume was made upto 50 μl using nuclease free water. The amplification was done using QB-96 (Quanta Biotech, UK) thermocycler using the following protocol: initial predenaturation at 95°C for 5 min, 35 cycles of denaturation at 94°C for 45 s, annealing at 53°C for 45 s, and extension at 72°C for 45 s followed by final extension at 72°C for 10 min. Until further processing, the PCR tubes were held at 4°C. The amplicon was subjected to agarose gel electrophoresis (1.5% gel) containing 1 μg/ml ethidium bromide. The gels were examined and photographed by gel documentation system.
Dry-reagent polymerase chain reaction mix for DNA
The DNA extracted from control and test strains were used for evaluation of the dry-reagent PCR mix. The ready to use dry-reagent PCR mix tubes were obtained from M/s Bhat Biotech India Pvt. Ltd., Bangalore. PCR was performed with addition of 2 μl of extracted DNA and 48 μl of PCR grade water to the dry-reagent PCR mix tubes, and the tubes were vortexed and centrifuged briefly and transferred to thermal cycler. PCR reactions involved initial predenaturation at 95°C for 5 min followed by thirty amplification cycles of denaturation at 94°C for 45 s, annealing at 53°C for 45 s, extension at 72°C for 45 s, and final extension at 72°C for 10 min.
Dry-reagent polymerase chain reaction mix for bacterial isolates
All the bacterial isolates, including test and control isolates, were evaluated by dry-reagent PCR mix. Ready to use dry-reagent PCR mix tubes were provided by M/s Bhat Biotech India Pvt. Ltd., Bangalore. To the dry-reagent PCR mix tube, 5 μl of fresh bacterial growth (LB broth inoculated with single colony of pure growth on brain heart infusion agar and incubated overnight at 37°C) and 45 μl of PCR grade water were added. The tubes were vortexed, centrifuged briefly, and placed in the thermal cycler for amplification. The protocol for amplification was initial predenaturation at 95°C for 8 min followed by 30 cycles of denaturation at 94°C for 45 s, annealing at 53°C for 45 s and extension at 72°C for 45 s and a final extension at 72°C for 10 min.
Results
In both conventional wet-reagent mix and dry-reagent PCR mix, the 100 clinical samples containing Acinetobacter had amplified specific PCR product [Table 1] which was comparable with the A. baumannii (ATCC) culture [Figures 1 and 2]. To evaluate the sensitivity and specificity of our primer, other 100 non-Acinetobacter isolates were tested by all three methods of PCR considering conventional wet-reagent PCR mix as gold standard. All the other bacterial strains showed negative for rpoB Acinetobacter primer [Table 2]. From the result, it was inferred that the PCR reaction was 100% sensitive and specific to the target Acinetobacter.

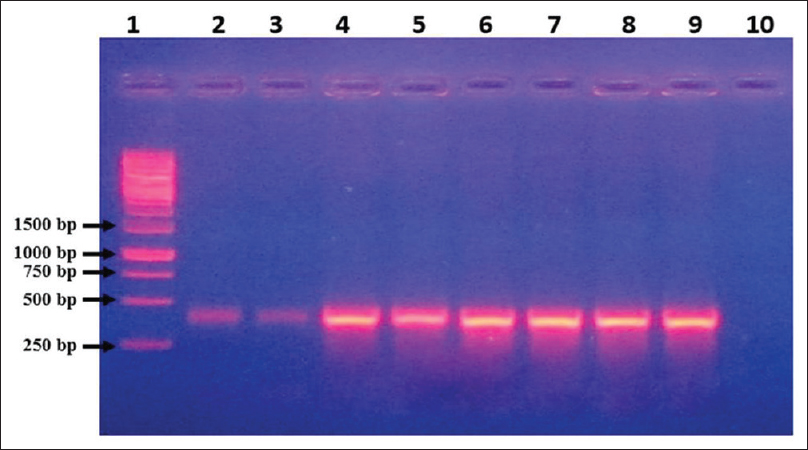
- Agarose gel electrophoresis of DNA amplified using conventional wet-reagent mix polymerase chain reaction with rpoB gene primers. Lane 1: Molecular size marker (1 kb Ladder), Lane 2–3: Extracted DNA of Acinetobacter baumannii (ATCC) used as template, Lane 4–9: Extracted DNA from different clinical isolates of Acinetobacter used as template, Lane 10: Blank
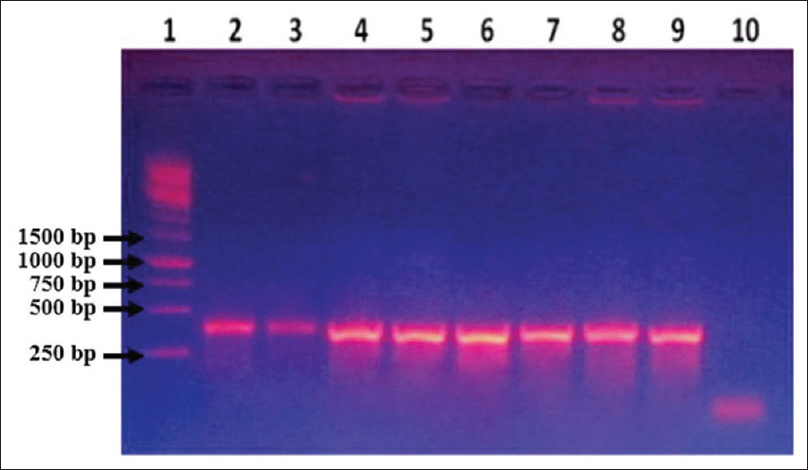
- Agarose gel electrophoresis of polymerase chain reaction products obtained from Acinetobacter using dry-reagent mix polymerase chain reaction with rpoB primers. Lane 1: Molecular size marker (1 kb Ladder), Lane 2–3: Extracted DNA of Acinetobacter baumannii (ATCC) used as template, Lane 4–5: Acinetobacter baumannii (ATCC) overnight grown culture as template, Lane 6–7: DNA extracted from clinical isolate of Acinetobacter as template, Lane 8–9: Overnight grown culture of Acinetobacter clinical isolate as template, Lane 10: Blank

Discussion
PCR is a well-known technique for the detection and identification of pathogens in clinical laboratories.[7] In spite of high sensitivity and specificity, this test is not widely used in the undeveloped countries due to paucity of resources, expertise, and high cost.
Dry-reagent PCR mix is, hitherto, scarcely used technique in spite of having distinctive advantage. It has enormous potential and applicability in clinical laboratories and successfully employed for the rapid detection of pathogens. Stabilized, freeze-dried dry PCR was used for the detection of Mycobacterium spp.[11] Dry PCR reaction mix was used for the detection of an endemic condition called buruli ulcer caused by Mycobacterium ulcerans. They have compared with other standard methods available and reported that there is 30% increase in positivity ratio.[1314] Quantitative dry PCR for the detection of endemic infection by Yersinia pestis was developed using a mixture of carbohydrates (trehalose and dextran) to obtain high stability.[15] A rapid thermostabilized triplex PCR for the identification of toxigenic and nontoxigenic strains of V. cholerae was developed which required minimum pipetting steps and was cold chain free. PCR reagents and the specific primers were lyophilized into a pellet form in the presence of trehalose, which acts as an enzyme stabilizer. The triplex PCR was validated with 174 bacteria-spiked stool specimens and was found to be 100% sensitive and specific.[16] Development of freeze-dried (lyophilized) reagents that do not require cold chain, with sensitivity at the level of wet reagents for the detection of avian influenza virus has brought on-site remote testing to a practical goal.[8] Dry-reagent-based PCR was developed for the rapid on-site detection of microbial pathogens such as blackleg of ruminants caused by Clostridium chauvoei. Basic PCR reagents (bovine serum albumin, PCR buffer, MgCl2, and primers) were dried on polyolefin matrices, showed stability at ambient temperatures for up to 10 months without any loss of functionality, eliminated PCR error, and saved time.[6]
We have used our proprietary method of producing dry-reagent mix that does not use lyophilization or any expensive matrices and therefore cost-effective. Moreover, a commercial DNA polymerase resistant to inhibitors was used in the preparation of the mix which eliminates the need for DNA isolation. The results showed that dry-reagent PCR mix showed 100% sensitivity and specificity against conventional wet-reagent PCR mix as gold standard [Tables 1 and 2]. All the 100 phenotypically identified Acinetobacter isolates showed the PCR amplified DNA band of rpoB gene at 397 bp region confirming the identity of the isolates, while set of 100 non-Acinetobacter bacterial isolates were negative by all the PCR methods used.
The hallmark of the dry-reagent PCR mix was elimination of the DNA extraction step before amplification. It was achieved by proprietary technique of processing the samples. The mix aimed at breaking the bacterial cells and dissolving the proteins, lipids, and other cell debris. The use of robust commercial polymerase helped to evade the inhibitors. The results indicate that the dry-reagent PCR mix was able to amplify the target sequence without any inhibition as the sensitivity recorded was 100%. Our results also show that either the use of isolated DNA or whole bacterial cells for dry-reagent PCR mix did not influence the sensitivity or specificity of the test. Elimination of the step of DNA extraction has multiple advantages. This obviously will reduce the chances of contamination. It saves time as manual extraction of DNA takes 24–48 h. DNA obviates the need to store and skillfully handle several sensitive reagents essential in the process of extraction. The handling becomes easy, making the assay user-friendly.
As the dry-reagent mix is distributed in PCR tubes, and the cell suspension is added just before running PCR, there is no need to make a master mix before the assay. Looking at the ease, speed, robust nature, reliability, and cost-effectiveness, this technique has tremendous potential in the identification of culture isolates. This method has also been tested for identification of E. coli isolates successfully at our center (unpublished data).
There is a need to assess this technique for diagnosis of infections directly from clinical samples. Dry-reagent PCR mix will be a boon, especially in the management of critically ill patients where speed and reliability of the test result will be a key in successful therapy of infections.
Financial support and sponsorship
All the authors are grateful for the financial assistance provided by Bhat Biotech India Pvt. Ltd., for the project.
Conflicts of interest
There are no conflicts of interest.
References
- A PCR-based method to differentiate between Acinetobacter baumannii and Acinetobacter genomic species 13TU. Clin Microbiol Infect. 2007;13:1199-201.
- [Google Scholar]
- High rates of resistance to colistin and polymyxin B in subgroups of Acinetobacter baumannii isolates from Korea. J Antimicrob Chemother. 2007;60:1163-7.
- [Google Scholar]
- Prevalence of OXA-type carbapenemase genes and genetic heterogeneity in clinical isolates of Acinetobacter spp. from Mangalore, India. Microbiol Immunol. 2011;55:239-46.
- [Google Scholar]
- Validation of partial rpoB gene sequence analysis for the identification of clinically important and emerging Acinetobacter species. Microbiology. 2009;155:2333-41.
- [Google Scholar]
- Sequencing of the rpoB gene and flanking spacers for molecular identification of Acinetobacter species. J Clin Microbiol. 2006;44:827-32.
- [Google Scholar]
- Dry-reagent-based PCR as a novel tool for the rapid detection of Clostridium spp. J Med Microbiol. 2013;62:1588-91.
- [Google Scholar]
- Rapid diagnosis of avian influenza virus in wild birds: Use of a portable rRT-PCR and freeze-dried reagents in the field. J Vis Exp 2011:pii: 2829.
- [Google Scholar]
- Rapid detection of methicillin-resistant Staphylococcus aureus by a newly developed dry reagent-based polymerase chain reaction assay. J Microbiol Immunol Infect. 2014;47:484-90.
- [Google Scholar]
- Single-tube nested PCR with room-temperature-stable reagents. Genome Res. 1995;4:376-9.
- [Google Scholar]
- Stabilized, freeze-dried PCR mix for detection of mycobacteria. J Clin Microbiol. 1998;36:1798-800.
- [Google Scholar]
- Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1989.
- Dry-reagent-based PCR as a novel tool for laboratory confirmation of clinically diagnosed Mycobacterium ulcerans-associated disease in areas in the tropics where M. ulcerans is endemic. J Clin Microbiol. 2005;43:271-6.
- [Google Scholar]
- Dry reagent-based polymerase chain reaction compared with other laboratory methods available for the diagnosis of Buruli ulcer disease. Clin Infect Dis. 2007;45:68-75.
- [Google Scholar]
- Ambient stable quantitative PCR reagents for the detection of Yersinia pestis. PLoS Negl Trop Dis. 2010;4:e629.
- [Google Scholar]
- Development of a dry reagent-based triplex PCR for the detection of toxigenic and non-toxigenic Vibrio cholerae. J Med Microbiol. 2011;60:481-5.
- [Google Scholar]



