
Translate this page into:
Griscelli Syndrome: Hemophagocytic Lymphohistiocytosis with Silvery Hair
Address for correspondence: Dr. Prashant B Mahalingashetti, Email: pmshetti49@yahoo.co.in
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Fever is associated with multitude etiologies and its diagnosis often requires number of investigations. Of interest, are certain rare entities like Hemophagocytic Lymphohistiocytosis (HLH). Seventeen year female patient presented with fever and chills since a week. She was the first child of a consanguineous marriage. Physical examination revealed pallor, icterus, hepato-splenomegaly, cervical lymphadenopathy and white hair with silvery sheen on scalp, eyebrows and eyelashes [Figure 1]. The routine investigations suggested pancytopenia with Hemoglobin-5.8 gm/dl, White cells-2,180/μL, Platelets-33,0000/μL. Peripheral smear showed no parasites. Serological tests for dengue, typhoid and leptospirosis turned negative. Serum biochemical parameters showed bilirubin-3.3 mg/dl, direct bilirubin-0.3 mg/dl, Alkaline phosphatase-276U/L, Triglycerides-164 mg/dl and LDH-2250U/L. No bacterial or mycobacterial growth was seen on blood culture. Aspiration smears of cervical lymph nodes showed features of reactive lymphadenitis. The patient was referred for bone marrow evaluation. Marrow smears showed hemophagocytosis [Figures 2a–d]. No pathogens were identified on smears. Review of all clinical and laboratory data suggested the diagnosis of Griscelli syndrome (GS) type 2.
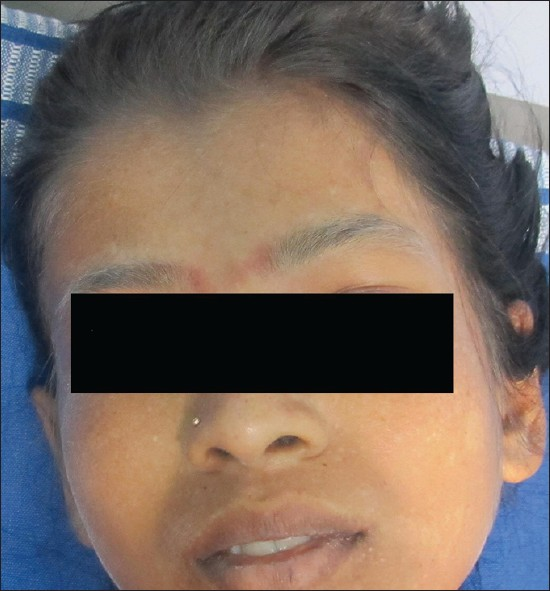
- Silvery white hair on forehead and eyebrows
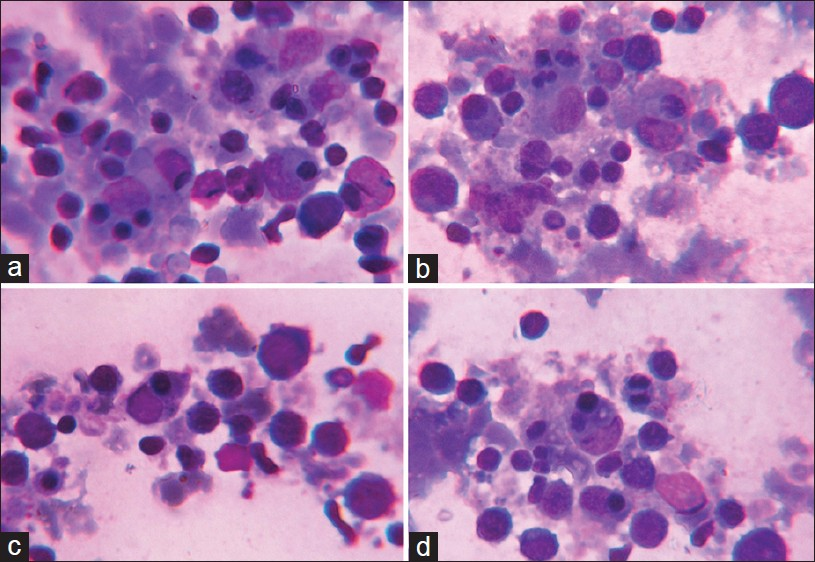
- (a-d) Phagocytosis of erythroid precursors in bone marrow smears (Leishman stain, oil immersion ×1000)
The case presented here sufficed the diagnostic criteria for HLH laid by Histiocytic Society which includes fever, hepatosplenomegaly, cytopenia involving at least two cell lineage, hypertriglyceridemia and/or hypofibrinogenemia coupled with hemophagocytosis in bone marrow, spleen or lymph node.[1]
HLH is classified as primary and secondary. Secondary HLH is seen associated with wide variety of diseases such as viral infection, lymphomas, solid organ malignancies and autoimmune disorders.[12] Genetic disorders like Familial HLH, Chediak Higashi syndrome, X linked lymphoproliferative disease and GS type 2 underlie primary HLH.[2] Primary HLH are usually diagnosed in young, rarely they are recognized at later age as in our case.
GS type 2 is caused by mutation in RAB27A gene, and presents as hypomelanosis recognizable by the characteristic silvery sheen of hairs, variable immunological impairment, an accelerated phase of hemophagocytosis, with or without neurological impairment.[23] Differential diagnosis include other primary HLH. Giant cytoplasmic granules in leucocytes are evident in Chediak Higashi syndrome; while X linked lymphoproliferative disease and Familial HLH do not exhibit albinism.[34]
Thus, role of bone marrow examination in diagnosis of fever is irrefutable. Presence of hemophagocytosis should prompt a battery of tests to rule out secondary causes. Physical examination and simple tests like blood smear evaluation can help differentiate among various genetic diseases causing primary HLH.
REFERENCES
- Hemophagocytic lymphohistiocytosis: Overview and diagnostic procedure.A case induced by an expansion of monoclonal EBV-negative. NK cells. Inmunologia. 2009;28:135-46.
- [Google Scholar]
- Griscelli Syndrome. 2012. Available from: http://www.orpha. net/data/patho/GB/uk.griscelli.pdf
- [Google Scholar]



