
Translate this page into:
Mutations in SARS-CoV-2 Leading to Antigenic Variations in Spike Protein: A Challenge in Vaccine Development

Address for correspondence Sarman Singh, MD, FRSC, FRCP, Director, All India Institute of Medical Sciences, Bhopal, 462020, Madhya Pradesh, India (e-mail: sarman_singh@yahoo.com, sarman.singh@gmail.com).
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Objectives
The spread of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) virus has been unprecedentedly fast, spreading to more than 180 countries within 3 months with variable severity. One of the major reasons attributed to this variation is genetic mutation. Therefore, we aimed to predict the mutations in the spike protein (S) of the SARS-CoV-2 genomes available worldwide and analyze its impact on the antigenicity.
Materials and Methods
Several research groups have generated whole genome sequencing data which are available in the public repositories. A total of 1,604 spike proteins were extracted from 1,325 complete genome and 279 partial spike coding sequences of SARS-CoV-2 available in NCBI till May 1, 2020 and subjected to multiple sequence alignment to find the mutations corresponding to the reported single nucleotide polymorphisms (SNPs) in the genomic study. Further, the antigenicity of the predicted mutations inferred, and the epitopes were superimposed on the structure of the spike protein.
Results
The sequence analysis resulted in high SNPs frequency. The significant variations in the predicted epitopes showing high antigenicity were A348V, V367F and A419S in receptor binding domain (RBD). Other mutations observed within RBD exhibiting low antigenicity were T323I, A344S, R408I, G476S, V483A, H519Q, A520S, A522S and K529E. The RBD T323I, A344S, V367F, A419S, A522S and K529E are novel mutations reported first time in this study. Moreover, A930V and D936Y mutations were observed in the heptad repeat domain and one mutation D1168H was noted in heptad repeat domain 2.
Conclusion
S protein is the major target for vaccine development, but several mutations were predicted in the antigenic epitopes of S protein across all genomes available globally. The emergence of various mutations within a short period might result in the conformational changes of the protein structure, which suggests that developing a universal vaccine may be a challenging task.
Keywords
SARS-CoV-2
spike protein
mutations
epitopes
antigenicity

Introduction
Since the rapid outbreak of 2019 novel coronavirus (2019-nCoV, later named SARS-CoV-2 or severe acute respiratory syndrome coronavirus 2) in Wuhan, China, the World Health Organization on January 30, 2020, declared the SARS-CoV-2 epidemic as a public health emergency of international concern. The enduring pandemic has caused nearly 5 million detected cases of coronavirus disease 2019 (COVID-19) illness and claimed over 3,25,563 lives worldwide as of May 20, 2020 according to COVID-19 Resource Center Johns Hopkins.1 However, so far, no proven therapeutic or effective vaccine candidate has been found.2
For developing a drug or vaccine, the protein profiling and/or genomic information of the pathogen is extremely crucial. To understand genetic landscape of SARS-CoV-2 virus, scientists have worked tirelessly and the complete genome sequences of virus isolates are published. Now, many isolates have been sequenced completely or partially and are available in the database for scientific community.3 It is found that the genome size of SARS-CoV-2 varies from 29.8 kb to 29.9 kb. Specific genetic characteristic in its genome have also been found.4 The genome consists of four structural proteins including spike protein (S), envelope (E), membrane (M), and nucleocapsid (N) proteins.3 Of the four glycoproteins, the M protein is reported to have role in determining the shape of the virus envelope and stabilizing the nucleocapsids, the N protein is involved in processes related to the viral genome, the viral replication cycle, and the cellular response of host cells to viral infections. The E protein which is the smallest protein in the SARS-CoV-2 structure plays the role in the production and maturation of this virus.4 However, the glycoprotein S is the core transmembrane monomer of approximately 180 kDa size with two subunits S1 and S2. This glycoprotein mediates membrane fusion and finally facilitates virus entry (receptor-binding and entry of virion into the target cells).5 The receptor binding domain (RBD) (residues 319–541) of the subunit S1 is known to interact with angiotensin-converting enzyme 2 (ACE-2), which provides tight binding to the peptidase domain of ACE-2.6 This gives an impression of RBD being an important element of virus–receptor interaction and has an essential role in virus-host range, tropism, and infectivity.
The RBD sequences of different SARS-CoV-2 strains that are circulating globally were initially thought to be conserved; however, with the availability of sequencing data several mutations have been reported.7 8 These findings emphasize the argument, that there may be correlation of the mutated strains circulating in a particular geographic setting with higher mortality rates and transmission patterns beside other combating factors.9 The mutation rate in ribonucleic acid (RNA) viruses is intensely high which can be million times higher than that of their hosts and this results in virulence modulation and evolutionary capability for better viral adaptation.7 Genetic depiction of virus mutations can thus offer valuable insights for assessing the fitness of drug resistance, immune escapism, and pathogenesis. Due to its receptor binding property, the S protein is also supposed to be immunogenic and a putative target for developing the neutralizing antibodies and vaccines. It is reported that single-point mutations in the conserved amino acid residues in the RBD region completely abolishes the capacity of full-length S protein to induce neutralizing antibodies.10 Thus, virus mutation studies can be crucial for designing new vaccines and antiviral drugs.
In this study, we aimed to predict the mutations in the spike protein (S) of SARS-CoV-2 genomes available in the database (whole genome sequences as well as partial coding sequences of spike protein) and analyze the effect of each mutation on the antigenicity of the predicted epitopes. This information may be helpful in predicting the transmission and infectivity of various SARS-CoV-2 strains circulating worldwide.
Materials and Methods
Selection of SARS-CoV-2 Datasets
Entrez Direct (EDirect) utilities were used to access the NCBI’s nucleotide database by using in-house developed bash scripts to batch download the data. We used query keyword or phrase as “severe acute respiratory syndrome coronavirus 2” and “spike coding sequences (CDS)” by applying ESearch and EFetch utilities implemented in bash scripts to download the dataset for genome and spike proteins that were available till May 1, 2020. A total of 1,325 complete draft genomic sequences of SARS-CoV-2 and additional 279 CDS having partial genomes coding spike protein (total 1,604 CDS of spike protein) were downloaded in FASTA format available globally from NCBI database (►Fig. 1).

- SARS-COV-2 genome datasets from different countries available on NCBI till May 1, 2020. SARS-COV-2, severe acute respiratory syndrome coronavirus 2.
Multiple Sequence Alignment and Correlation of Genomic SNPs and Protein Mutations
Multiple sequence alignment (MSA) of all the 1,325 complete genome sequences as well as 1,604 CDS was performed using ClustalW-MPI with default parameters.11 Generated SNPs were identified by in-house developed bash scripts to batch process the data using Blade Server (Dell PowerEdge FC640 Server) with 256 GB RAM and 40 Core processor with 2.30 GHz. After MSA, each genome and spike protein were marked based on the location and clustering was done based on 100% similarity for ease of visualization and analysis. Visualization was performed by using JalView 2.11.1.0.12
Construction of Phylogenetic Tree
The output SNP alignment generated from MSA was used to assemble a maximum likelihood phylogenetic tree using RAxML (Randomized Axelerated Maximum Likelihood RAxML 8.2.12).13 Phylogenetic trees were visualized using the Interactive Tree of Life (iTOL) V5 with their respective metadata.14
Prediction of Antigenic Epitopes
EMBOSS antigenic software was used to predict the antigenic regions and the epitopes in the 88 unique spike proteins based on antigenic scores using the formula:
Antigenic propensity column A(p) = f(Ag)/f(s)
where f(Ag) = antigenic frequency; f(s) = surface frequency and antigenic score ≥ 1.0 is considered potentially antigenic.15-17
The data for different epitopes were analyzed and the epitopes with high antigenicity were superimposed on the structure of spike protein.
Results
SNP Analysis of SARS-CoV-2 Genome Set
Overall, 1,197 SNPs were found in 1,325 complete genome datasets. On the basis of similarity these were classified in 782 clusters. However, among CDS of spike proteins (1,325 complete genomes and 279 partial CDS) a total 140 SNPs in 88 clusters were found. Further SNP analysis resulted in identification of SNPs in a gene stretch of 21,563–25,384 bp in the S gene, encoding the spike (S) protein. The most predominant SNP predicted in the gene encoding S protein was 23402A>G in 48.2% of overall genomes under study. In addition to several single mutations in the S gene of all available genomes, we also predicted double mutations such as 22436G>T, 22439C>G, 22444C>A, 22445C>T (corresponding to four amino acid antigenic drift ALDP -> SVES at position 292–295) and 21723T>G (L54W); 21726T>A (F55I) in two different genomes from the United States. List of all the SNPs in the RBD, antigenic sites, and double mutations among strains is shown in ►Table 1. One deletion (21994–21996delTTA) was also found in an Indian strain (MT012098.1). No copy number variants were observed in this virus.
| Genome ID | SNPs | Corresponding mutations | |
|---|---|---|---|
| 1 | MT263393_USA, MT385442_USA, MT370859_USA, MT370859_USA, MT371019_USA, MT334572_USA, MT334572_USA, MT334572_USA, MT345871_USA, MT358718_USA, MT326090_USA, MT396242_India, MT334540_USA, MT370991_USA, MT334558_USA, MT345818_USA, MT358689_USA, MT326092_USA, MT322423_USA, MT252781_USA, MT350253_USA, MT334547_USA, MT344952_USA, MT370923_USA, MT358637_India, MT259281_USA, MT345864_USA, MT252726_USA, MT334559_USA, MT385447_USA, MT370831_USA, MT259249, MT375449_USA, MT326126_USA, MT345831_USA, MT358669_USA, MT371011_USA, MT358745_USA, MT374108_Taiwan, MT394864_Germany, MT371569_CzechRepublic, MT371568_CzechRepublic | 23402A>G | D614G |
| 2 | MT375449_USA | 21726T>A | F55I |
| 3 | MT358745_USA | 22436G>T | A292S |
| 4 | MT358745_USA | 22439C>G | L293V |
| 5 | MT358745_USA | 22444C>A | D294E |
| 6 | MT358745_USA | 22445C>T | P295S |
| 7 | MT375449_USA | 21723T>G | L54W |
| 8 | MT385491_USA | 21723G>C | L54F |
| 9 | MT326041_USA | 21774C>T | S71F |
| 10 | MT263450_USA | 21893G>A | D111N |
| 11 | MT159716_USA | 22033C>A | F157L |
| 12 | MT358745_USA | 22441C>G | L293V |
| 13 | MT358745_USA | 22444C>A | D294E |
| 14 | MT326157_USA | 25311G>T | C1250Y |
| 15 | MT350270_USA | 23120G>T | A520Sa |
| 16 | MT263418_USA | 23004T>C | V483Aa |
| 17 | MT358726_USA | 22988G>A | G476Sa |
| 18 | MT246483_USA | 22592G>T | A344Sa,c |
| 19 | MT263457_USA | 23119T>A | H519Qa |
| 20 | MT259253_USA | 22604G>A | A348Ta |
| 21 | MT358689_USA | 23147A>G | K529Ea,c |
| 22 | MT370923_USA | 23125G>T | A522Sa,c |
| 23 | MT345818_USA | 22530C>T | A419Sa,b,c |
| 24 | MT370991_USA | 22530C>T | T323Ia,c |
| 25 | MT259281_USA | 24368G>T | D936Y |
| 26 | MT263420_USA | 25064G>C | D1168H |
| 27 | MT372482_Malaysia | 22437C>T | A292S |
| 28 | MT372482_Malaysia | 22439C>A, 22441T>G | L293V |
| 29 | MT372482_Malaysia | 22443A>T | D294E |
| 30 | MT372482_Malaysia | 22446C>A | P295S |
| 31 | MT372482_Malaysia | 22448C>T, 22450C>T | L296F |
| 32 | MT372482_Malaysia | 22452C>G, 22453A>G | S297W |
| 33 | MT358745_Malaysia | 22441C>A | L293M |
| 34 | MT358745_Malaysia | 22443A>T | D294I |
| 35 | MT396242_India | 25311G>A | C1250F |
| 36 | MT012098_India | 22782G>T | R408Ia |
| 37 | MT050493_India | 24353C>T | A930V |
| 38 | MT291835.2_China | 23521C>T | A653V |
| 39 | MT230904_Hongkong | 22661G>T | V367Fa,b,c |
Abbreviation: SNP, single nucleotide polymorphisms.
aSNPs and corresponding mutations in RBD regions.
bVariations in highly antigenic epitopes.
cNovel mutations.
Phylogenetic Analysis of Spike Protein
The alignment of 1,604 spike proteins extracted from 1,325 complete genomes and 279 partial CDS was performed and after clustering based on 100% identity, we identified 88 unique sequences with 88 hypervariable sites within these protein sequences. Based on the variable sites the phylogeny was inferred showing two major clades A and B with many subclades in the S protein of SARS-CoV-2 circulating worldwide (►Fig. 2).
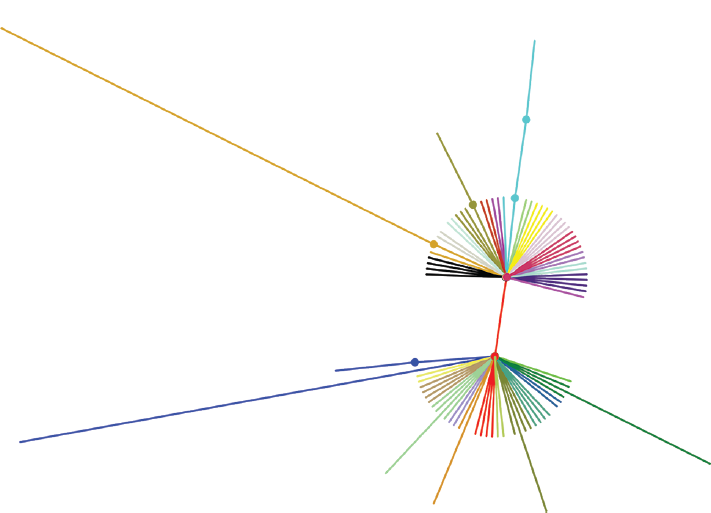
- Phylogenetic tree inferred from variable region of spike protein showing different clades by different colors circulating worldwide. Two Major clades A and B are formed due to the most prevalent mutations D614G. Other small subclades shown in different colors are due to different mutations amongst them. MT358745 and MT372482 are shown with different branch length due to antigenic drift of six amino acids (ALDPLS292VMIHFW) and four amino acid (ALDP292SVES), respectively.
Scoring of Antigenic Sites and Prediction of Spike Protein
Furthermore, the evaluation of the antigenicity of spike protein predicted 14 highest scoring antigenic epitopes (antigenic scores ≥ 1.0) due to variations in each (at positions L54F, L54W, F55I, S71F, D111N, F157L, L293M, L293V, D294E, D294I, A419S, V367F, A348V, and A653V). Out of these, amino acid changes were noted at positions A348V, V367F and A419S in the RBD with V367F and A419S being novel. Other speculated mutations in the putative epitopes lying within RBD showing less antigenicity were T323I, A344S, R408I, G476S, V483A, H519Q, A520S, A522S, and K529E out of which T323I, A344S, A522S, and K529E are also novel. In addition, regions outside RBD (4–19, 1,215–1,256, 1,039–1,071, 123–146, 607–629, 1,123–1,136, 857–867, 535–541) also infer high antigenicity (based on predicted antigenic score in the range 1.167–1.261) with variations in S protein of different genomes from similar locations as shown in ►Fig. 3. The antigenic epitopes are depicted in the protein structure of spike protein (►Fig. 4). Two mutations A930V and D936Y were observed in the heptad repeat domain 1 (HR1) and one mutation D1168H in heptad repeat domain 2 (HR2).

- Heat map of antigenic epitopes in spike protein showing antigenicity in different genomes. Left first column with purple color shows antigenicity for receptor binding domain (RBD) (Arg319-Phe541).

- Structural details of spike protein showing antigenic region and epitopes within the protein by different colors, (A) front view of the protein (B) view after rotation of 90 degrees.
Discussion
Several studies have shown that mutations within the spike protein influence virus–host interaction.18 Among the four proteins, viz., M, S, N and E, the M protein is known to play a significant role in virus assembly, role of E protein involves the production and maturation of this virus, the N protein is involved in processes related to viral replication cycle, and cellular response of host cells to viral infections, and S protein is the major target for neutralizing antibodies as it mediates the fusion and facilitates viral entry into host.4,5 In the present study, we found that although multiple genetic variants were identified in the same country, yet there were some unique mutations found in a particular country, which suggests that diversity of S protein mutations might have significant role in the pathogenicity of this virus in countries with high or low mortality rates, as proposed by others also.19 We predicted 140 SNPs in the S protein with A>G and vice versa in SARS-CoV-2 genomes submitted from India, T>A and C>T from China and the interchange of all four nucleotides (C>T, T>A, A>G, G>T, C>G, C>A, G>C, T>C, A>T, G>A) in genomes submitted from the United States. The data from the United States is significant. However, it might be because maximum genomes were submitted from the United States only. The SNP profile revealed that the S protein mutations were predominant at specific positions only. These mutations are expected to make the virus more capable to escape from the host immune and might help in natural selection and evolution of the SARS-CoV-2, as reported by Andersen et al.20 It is important to mention that double mutations in the S protein were found only in the strains from the United States but not in genomes from other regions. These double mutations probably could have helped in the increased virulence of the virus.21 It has also been noticed that the death toll is comparatively higher in the United States than in other regions included in the study.1 This might probably indicate that the prevalence of several mutated strains within the provinces would have either reduced or increased its severity. It may also help in understanding the antigenic and immunogenic changes but the correlation of mutation with regional virulence could not be established due to statistical imbalance of the available genomes in the database at the time the study was done. Moreover, extensive research is required to correlate the mutations with the severity of the disease and mortality.
Out of 88 SNP clusters, D614G was found in 34 (38.6%) SARS-CoV-2 genomes. The amino acid change in 23403A>G variant (p.D614G) involves a change of large acidic residue D (aspartic acid) into small hydrophobic residue G (glycine). This observation is important, as this large difference in both size and charge may help compromise the binding affinity of antibodies against S protein, due to electrostatic interactions in the tertiary structure of protein group. This may hinder the developments of vaccines and might potentiate the virus for antigenic drifts.22 The effect of deletion variant (figured in one SARS-CoV-2 genome from India) on the viral phenotypes needs further investigation. The high frequency of genetic mutations in RNA viruses is well known but in the genomes of SARS-CoV-2, we found a series of single amino acid variations. This can affect the virus evolution and emergence of the new strains.21 The mutations in the RBD found in our study predicted conformational changes in the S1 domain of spike protein. The mutations in RBD play an important role while designing new drugs, as suggested in a recent study.23 These mutations might affect the interaction of viral RBD with the host receptor. Our study revealed 12 mutations of which six were novel mutations (►Table 1). Out of the six novel mutations two were exhibiting high antigenicity while others were in the less antigenic region. The amino acid change observed in the antigenic epitopes were from positively charged to uncharged amino acids (R->I, H->Q), and negatively charged to uncharged (D->N, D->G, D->Y) amino acids. We also found mutations in negative to positively charged (D->H) amino acids. These replacements might influence the tertiary structure of the proteins and facilitate the increased virulence by escaping host immune response.24
The sequences in the HR1 (residues 902–952) and HR2 (residues 1,145–1,184) regions tend to form dimeric or trimeric helix bundles.25 As the S protein of coronavirus are homodimers or homotrimers, these HR regions may undergo oligomerization and result in the conformational change of S protein during virus–host cell fusion.26 These regions show different conformations in different fusion states and are known to be the most conserved among other regions in S protein of SARS-CoV.25,27 However, a previous study shows variations in HR1 domain which forms helical bundles with HR2 to facilitate fusion and entry of virus into the host and hypothesizes that the mutation A1168V in HR2 domain along with A930V mutation in HR1 domain confers peptide entry inhibitor resistance in mouse hepatitis coronaviruses.28 Hence the mutation A930V in HR1 domain and D1168H in HR2 domain found in our study might be relevant in explaining the pathogenesis of SARS-CoV-2.
With the rapid spread of this virus and limitation of specific therapy, studies are being focused on exploring the potential of neutralizing antibodies (as in plasma therapy) against vulnerable epitopes of S protein.29 Our study predicts some sequence-based epitopes conserved in all the spike proteins but with variable antigenicity (►Fig. 3).
Conclusion
Most of the vaccines strategies against COVID-2 are focusing on the predicted epitopes of SARS-CoV-2 spike protein. This protein is also proposed to be the most potent and specific drug target and for designing neutralizing antibodies. Our findings indicate that vaccine designing against SARS-CoV-2 could be a challenging task. Even though both RNA based, and peptide-based vaccines are being developed in more than seven laboratories, our observations may be useful in the efficacy analysis of these vaccine candidates.
Authors’ Contribution
P.K.S. contributed to the conceptualization, data curation, formal analysis, methodology, and original draft writing. U.K. contributed to the methodology, software, and original draft writing. S.B.R. contributed toward the methodology, resources, software, and original draft writing. S.R.M. worked toward the investigation methodology, resources, and original draft writing. S.S. did the conceptualization, funding acquisition, project administration, resources, supervision, writing review, and editing.
Conflict of Interest
P.K.S. and U.K. are research officers in a Department of Biotechnology funded unrelated project (BT/PR23016/NER/95/581/2017).
Funding
None.
Reference
- An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(05):533-534.
- [CrossRef] [PubMed] [Google Scholar]
- A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci. 2020;21(07):2657.
- [CrossRef] [PubMed] [Google Scholar]
- Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19:100682.
- [CrossRef] [PubMed] [Google Scholar]
- Coronavirus envelope protein: current knowledge. Virol J. 2019;16(01):69.
- [CrossRef] [PubMed] [Google Scholar]
- Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260-1263.
- [CrossRef] [PubMed] [Google Scholar]
- Structure of mouse coronavirus spike protein complexed with receptor reveals mechanism for viral entry. PLoS Pathog. 2020;16(03) e1008392
- [CrossRef] [PubMed] [Google Scholar]
- Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18(01):179.
- [CrossRef] [PubMed] [Google Scholar]
- Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12(03):254.
- [CrossRef] [PubMed] [Google Scholar]
- Real estimates of mortality following COVID-19 infection. Lancet Infect Dis. 2020;20(07):773.
- [CrossRef] [PubMed] [Google Scholar]
- Single amino acid substitutions in the severe acute respiratory syndrome coronavirus spike glycoprotein determine viral entry and immunogenicity of a major neutralizing domain. J Virol. 2005;79(18):11638-11646.
- [CrossRef] [PubMed] [Google Scholar]
- ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics. 2003;19(12):1585-1586.
- [CrossRef] [PubMed] [Google Scholar]
- Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(09):1189-1191.
- [CrossRef] [PubMed] [Google Scholar]
- RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688-2690.
- [CrossRef] [PubMed] [Google Scholar]
- Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256-W259.
- [CrossRef] [PubMed] [Google Scholar]
- EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(06):276-277.
- [CrossRef] [PubMed] [Google Scholar]
- A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1-2):172-174.
- [CrossRef] [PubMed] [Google Scholar]
- New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25(19):5425-5432.
- [CrossRef] [PubMed] [Google Scholar]
- Potential therapeutic targeting of coronavirus spike glycoprotein priming. Molecules. 2020;25(10):e2424.
- [CrossRef] [PubMed] [Google Scholar]
- SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract 2020 DOI: 10.1111/ijcp.13525. [epub ahead of print]
- [CrossRef] [PubMed] [Google Scholar]
- The proximal origin of SARS-CoV-2. Nat Med. 2020;26(04):450-452.
- [CrossRef] [PubMed] [Google Scholar]
- Why are RNA virus mutation rates so damn high? PLoS Biol. 2018;16(08) e3000003
- [CrossRef] [PubMed] [Google Scholar]
- Electrostatic interactions in protein structure, folding, binding, and condensation. Chem Rev. 2018;118(04):1691-1741.
- [CrossRef] [PubMed] [Google Scholar]
- Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: a computational biology approach. Infect Genet Evol. 2020;84 104389
- [CrossRef] [PubMed] [Google Scholar]
- Lineage-specific differences in the amino acid substitution process. J Mol Biol. 2010;396(05):1410-1421.
- [CrossRef] [PubMed] [Google Scholar]
- Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363(9413):938-947.
- [CrossRef] [PubMed] [Google Scholar]
- Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017;114(42):11157-11162.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777-810.
- [CrossRef] [PubMed] [Google Scholar]
- Coronavirus escape from heptad repeat 2 (HR2)-derived peptide entry inhibition as a result of mutations in the HR1 domain of the spike fusion protein. J Virol. 2008;82(05):2580-2585.
- [CrossRef] [PubMed] [Google Scholar]
- A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020;368(6491):630-633.
- [CrossRef] [PubMed] [Google Scholar]



