
Translate this page into:
Prevalence and Impact of HMOX1 Polymorphism (rs2071746: A > T) in Indian Sickle Cell Disease Patients
 , Jasmita Dass1, Seema Tyagi1, Tulika Seth1, Renu Saxena1, Manoranjan Mahapatra1,
, Jasmita Dass1, Seema Tyagi1, Tulika Seth1, Renu Saxena1, Manoranjan Mahapatra1,
Address for correspondence: Manoranjan Mahapatra, MD, Department of Hematology-Room number-205, IInd floor, New Private Ward, All India Institute of Medical Sciences (AIIMS), New Delhi, 110029, India (e-mail: mrmahapatra@hotmail.com).
-
Received: ,
Accepted: ,
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Introduction
Fetal hemoglobin (HbF) levels play significant role in lowering down the morbidity and mortality in sickle cell disease (SCD) patients. Coinheritance of heme oxygenase-1 (HMOX1) rs2071746:A > T polymorphism may contribute to variable HbF levels in Indian SCD patients.
Objective
This study was aimed to evaluate the role of HMOX1 polymorphism and its impact on HbF level in Indian SCD patients.
Materials and Methods
One-hundred twenty confirmed cases of SCD and 50 healthy controls were recruited. Their mean age was 11.5 ± 8.6 years (range: 3–23 years). Quantification of Hb, HbA2, HbF, and HbS was done by capillary zone electrophoresis. Allele-specific polymerase chain reaction was used to genotype HMOX1 (rs2071746:A > T) gene polymorphism.
Results
Out of the 120 cases of SCD, 65 were hemoglobin sickle-shaped (HbSS) and 55 were sickle-beta thalassemia (Sβ). Out of 65 HbSS patients, 29 (44.6%) were heterozygous (AT), 20 (30.76%) were homozygous (TT), and 16 (24.61%) were found wild-type (AA) genotype. Out of 55 Sβ, 22 (40%) were heterozygous, 18 (32%) were homozygous and 15 (28%) were wild-type. Patients carrying HMOX1 (rs2071746:A > T), AT, and TT genotypes had less anemia, painful crisis, splenomegaly, hepatomegaly, jaundice, and blood transfusion. HbF level was found higher in TT genotype (in HbSS the HbF levels was 25.1 ± 4.4; in sickle-beta thalassemia the HbF levels was 36.1 ± 4.7) than wild-type(AA) and was statistically significant (p-value <0.001).
Conclusion
The TT genotype of the rs2071746:A > T polymorphism was associated with increased levels of Hb F (p < 0.001). It can serve as a HbF modifier in Indian sickle cell diseases patients.
Keywords
HMOX1
HPLC
polymorphism
HbF
SCD

Introduction
Sickle cell disease (SCD) is a chronic lifelong debilitating disorder that begins in childhood and characterized by point mutation in the β-hemoglobin gene (HBB), resulting in substitution of adenine for thymine (GAG > GTG) at codon 6 of the β-globin gene [NM_000518.5 (HBB):c.20A > T (p.Glu7Val)]. This base pair substitution leads to change in amino acid valine for glutamic acid that results in polymerization of the abnormal sickle-shaped hemoglobin (HbSS)polymers, that is, sickle hemoglobin (HbS) especially when hemoglobin is in deoxygenated state. As the disease progresses for longer duration, there is permanent deformation of the shape of initially reversibly sickled red blood cells into the rigid irreversibly sickle cells that cannot pass through the microcirculation efficiently, resulting in anemia, cell adhesion, vaso-occlusion, and ultimately painful sickle cell crisis.[1,2] The sickle cell gene is widespread across in India and it is believed that the occurrence of the HbS mutation in India is different and separate from those in Africa. This is known as the Asian haplotype.[3]
Among the predictors of survival in SCD, fetal hemoglobin (HbF) levels play very important and significant role in these patients as targeted therapy with an aim to raise the HbF levels may be one of the foundation stone in lowering down the morbidity and mortality. HbF inhibits HbS polymerization and its abundance in the red blood cells dilutes down the concentration of HbS, thus leading to amelioration of the symptoms.[4–6] Different studies have identified that high levels of HbF have a protective role by reducing the severity of SCD. HbF is present at residual levels in healthy adults with over 20-fold possible quantitative variation and studies have shown that 89% of this variation is heritable, although the genetic etiology for this is complex.[7,8]
A wide range of complications has been reported in SCD inducing microvascular obstruction, abnormal adhesion of leukocytes and platelets, inflammation, and hypercoagulation. The sickle hemoglobin gene haplotype observed among various tribal populations in Central and Southern India is similar to that of the Asian-Indian haplotype and is associated with high HbF levels.[9]
At present about 05% of the world's population are carriers of any one of the hemoglobin gene mutation, with predominance of heterozygous condition. Every year about 300,000 infants worldwide are born with thalassemia syndrome (30%) and sickle cell anemia (SCA) (70%). Although the percentage of carriers of thalassemia is greater than that of carriers of SCA globally, but because of the higher frequency of the sickle cell gene in certain regions, overall number of affected population is higher than that with thalassemia. Although the general incidence of β-thalassemia trait and SCD varies between 3 to 17% and 1 to 44%, higher consanguinity, caste and area endogamy result in some communities exhibiting greater incidence of SCD, thus making this disease a major public and genetic health problem in India.[10,11]
As some genes in the β-globin gene cluster can affect disease pathology, other genes outside the gene cluster such as heme oxygenase-1 (HMOX1) and chemokine receptor 5 (CCR5) can modulate disease severity and subsequently reduce the severity of clinical events such as vascular occlusion, pain, chest syndrome, priapism, leg ulcers, and hepatobiliary complications.[12–14]
HMOX1 gene, present on the long arm of chromosome 22, codes the heme oxygenase 1 (HO-1) enzyme, which acts in the breaking of the porphyrin ring of the heme to produce biliverdin and carbon monoxide.[15] The HMOX1 gene is over expressed in sickle cell patients.[8] Few studies have shown that polymorphisms of the HMOX1 gene, including rs2071746: A > T, cause an increase in the HbF level possibly lowering the disease severity. HO-1 also acts as a potential anti-inflammatory and cytoprotective enzyme that is activated by inflammation, hypoxia and ischemia, and plays an important role in establishment of hemostasis.[16] The elevated oxidative stress in SCD is attributed to loss of HO-1 function because of HMOX1 polymorphisms that lead to excess of free heme in plasma, that auto-oxidizes leading to superoxide formation which results in free radical mediated inflammatory cascade ultimately augmenting the complications.[17]
After thorough search in literature, we found that there have been very few studies that have addressed the effect of HMOX1 polymorphisms (HMOX1-413A/T (rs2071746) in SCD patients and hence, we planned this study to understand the prevalence of this polymorphism and its effect on HbF levels in Indian SCD patients.
Materials and Methods
Cases and Controls Selection
A total of 120 individuals with SCD were recruited at Clinical Hematology OPD of tertiary care center in North India between 2016 and 2020. Their mean age was 11.91 ± 9.0 years (range: 3–23 years). Complete history including detailed transfusion history, age at onset of disease, incidence of vaso-occlusive episodes, history of jaundice, and other complications of SCD was recorded as per the clinical proforma. Written informed consent was taken from all the patients and controls and ethical approval was obtained from the institutional ethics committee, before the conduct of this study.
Laboratory Testing
Venous blood (5 mL) was drawn in EDTA vial and complete blood count was measured by automated cell analyzer (SYSMEX K-4500, Kobe, Japan). Giemsa-stained peripheral blood smears were examined for red cell morphology. Serum bilirubin levels were measured using a standard laboratory method. Abdominal ultrasound was performed for the identification of gallstones. Capillary zone electrophoresis (SEBIA-PARK TECHNOLOGY, Lisses, France) was used for the quantitative assessment of hemoglobin variants HbS, HbA, HbF, and HbA2. Cases of compound heterozygosity for heterozygous Hbs & HBC, heterozygous Hbs & HbD, and heterozygous Hbs & HbE cases were excluded.
Molecular Analysis
Genomic DNA was isolated from peripheral blood leukocytes using the Bioserve DNA isolation kit. Confirmation of the sickle mutation (codon 6, GAG > GTG) was done by amplification refractory mutation system-polymerase chain reaction (ARMS-PCR). The primer sequences were as follows:
WT-AS (5′-ATG GTG CAC CTG ACT CCT GA-3′) WT- Fw control
CP517 (5′-CCC CTT CCT ATG ACA TGA ACT-3′) Rw control
MUT-AS (5′-CAG TAA CGG CAG ACT TCT CCA-3′) Fw mutant
MUT-CP267 (5′-GGG TTT GAA GTC CAA CTC CTA-3′) Rw mutant
Screening of HMOX1-413A/T (rs2071746) SNP was performed using allele-specific PCR. Based on the nucleotide sequence of GenBank S58267, two sets of primer pairs were designed to amplify a 307-bp target sequence that shares the same forward primer but differs only in the 3′-nucleotide end of the reverse primers, making them allele-specific. The primers and their sequence used for detection were one common forward and two reverse primers for the wild-type genotype (AA) and for the variant genotype (TT) as:
Forward primer (F): 5′-ACTGGCACTCTGCTTTATGTGTGA-3′
Reverse variant primer (R V): 5′-GGAGGCAGCGCTGCTCAGAGTAAT-3′
Reverse wild-type primer (R W): 5′-GGAGGCAGCGCTGCTCAGAGTAAA-3′
The conditions of amplification for the polymorphism (rs2071746: A > T) in the HMOX1 gene were as follows: initial denaturation at 95°C for 5 minutes, followed by 35 cycles at 95°C for 1 minute, annealing of primers in (62°C) for 1 minute, and extension at 72°C for 1 min., ending with a final extension at 72°C for 10 minute. The product size was 307bp.
Statistical Analysis
Data was expressed as mean ± standard deviation, Yates' chi-squared test was used to assess intergroup significance, and test was performed on EpiInfo statistics software (Version 3.5.1). Student's t-test was used to compare the means of groups using Graph-Pad software. The allelic and genotypic frequency was calculated. Allele frequency was calculated as the number of occurrences of the test allele divided by the total number of alleles. The differences among the HbF levels within the genotypes of polymorphism were calculated by analysis of variance (more than 2 groups were involved), using Graph Pad Prism version 3.06. A p-value less than 0.05 was taken as statistically significant.
Results
A total 120 SCD cases and 50 healthy controls were recruited. Out of the 120 cases of SCD, 65 (41 male and 24 female) cases with mean age of 11.91 ± 9.0 (range: 3–23 years) were HbSS, and 55 (34 male and 21 females) cases with mean age of 8.80 ± 7.87 (range: 3–19 years) were compound heterozygous for sickle-beta thalassemia (HbSβ). The mean age of controls was 11.5 ± 7.78 (range: 10–35 years). The hematological profile of cases and controls was given in ►Table 1. Most of the cases of HbSS had normocytic, normochromic anemia and those of HbSβ were microcytic, hypochromic anemia. Of the 65 patients of HbSS, 34 (53%) had mild clinical presentation, whereas 31(48%) had a severe clinical presentation with a history of vaso-occlusive crises and chest syndrome.
| Parameters | Normal range | SCD (n = 120) (mean ± SD, range) | Controls (n = 50) (mean ± SD, range) | |
|---|---|---|---|---|
| HbSS (n = 65) | HbSβ (n = 55) | |||
| Age | – | 11.91 ± 9.0, 3–23 | 8.80 ± 7.87, 3–19 | 11.5 ± 7.78, 10–35 |
| Sex, | ||||
| a. Males | – | 41 | 34 | |
| – | ||||
| RBC (millions cells/µL) | 4.2–5.4 | 3.3 ± 1.5, 1.9–4.5 | 3.2 ± 1.5,1.7–4.7 | 4.60 ± 0.48, 4.1–5.8 |
| Hb (g/dL) | 13–16.5 | 8.04 ± 2.45, 5.5–10.5 | 9.49 ± 1.4, 8.0–10.80 | 13.2 ± 1.48, 11.0–14.5 |
| MCV (fL) | 80–94 | 62.2 ± 4.9, 57–67. | 70.83 ± 5.8,65.03–76.6 | 82.4 ± 4.72, 77.6–87.1 |
| MCH (pg) | 27–31 | 26.45 ± 5.45, 21–31.9 | 33.33 ± 4.46,28.87–37.7 | 27.4 ± 2.30, 29.7–25.1 |
| MCHC (g/dL) | 33–37 | 31.16 ± 2.25,28.0–33.0 | 31.45 ± 3.53,27.9–34.9 | 35.1 ± 1.3, 33.8–36.4 |
| HbA2 (%) | 1.5–3.5 | 3.4 ± 1.8, 1.7–5.0 | 5.2 ± 0.7, 4.2–5.9 | 2.7 ± 0.44, 2.2–3.1 |
| HbF (%) | <0.6 | 19.46 ± 4.23,15.2–23.6 | 28.53 ± 4.86,23.6-33.3 | 0.69 ± 0.30, 0.34–0.98 |
| HbS (%) | – | 74.5 ± 6.5, 65.0–80 | 64 ± 6.2, 56–72 | – |
Abbreviations: HbF, fetal hemoglobin; HbSβ, sickle-beta thalassemia; HbSS, hemoglobin sickle shaped; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; RBC, red blood cell; SCD, sickle cell disease; SD, standard deviation.
HMOX1 polymorphism was studied in all subjects. Out of 65 HbSS patients, 29 (44.61%) patients were heterozygous (AT), 20 (30.76%) were homozygous (TT), and 16 (24.61%) were found wild-type (AA) genotype. Out of 55 HbSβ heterozygous patients, 22 (40%) were heterozygous, 18 (32%) were homozygous, and 15 (28%) were wild type. The incidence of AA, AT, and TT genotype was 20(36%), 27(50%), and 8(14%) in controls.
A comparison of hematological parameters in different genotypes of HMOX1 polymorphism for HbSS and HbSβ is given in ►Tables 2 and 3, respectively. The mean HbF level was significantly higher in AT and TT genotypes when compared to AA (wild-type) genotype both in HbSS and HbSβ cases. The frequency of HMOX-1 polymorphism was higher in among sickle homozygous than sickle-beta thalassemia patients. The impact of genotypes on the clinical features is shown in ►Fig. 1. Clinical severity was improved with variant genotype in sickle cell anemia as well as sickle-beta patients. Painful crisis, jaundice, gall stone, and blood transfusion were lesser in mutant variant than nonvariant. HbF level was found higher in variant (TT) genotype than wild-type and was statistically significant (p-value <0.001). ►Tables 4 and 5 depict the allelic frequency of HbSS and HbSβ cases.
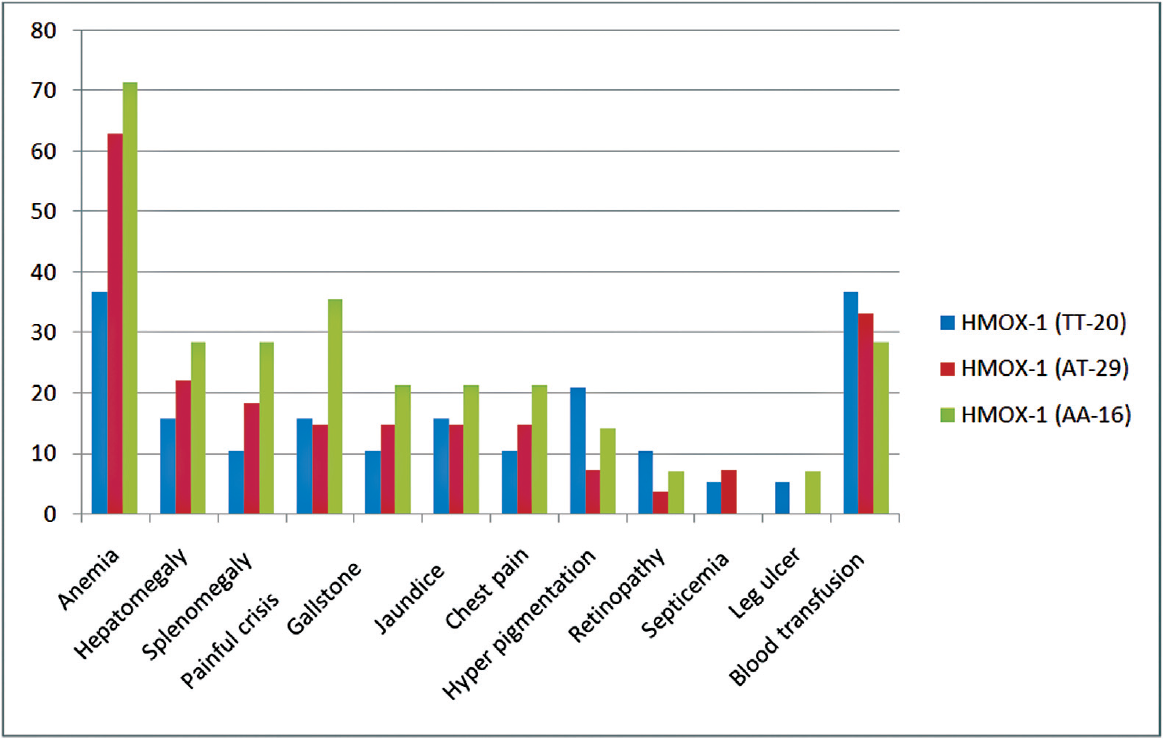
- Comparative clinical parameter with carrier and noncarrier heme oxygenase-1 (HMOX-1) in hemoglobin sickle shaped (n = 65).
| Hematological parameters | HMOX1(TT) n= 20 | HMOX1(AT) n = 29 | HMOX1(AA) n = 16 | p-Value |
|---|---|---|---|---|
| RBC (millions of cells/µL) | 3.8 ± 2.2 | 3.1 ± 1.8 | 3.5 ± 1.2 | 0.001 |
| Hb (g/dL) | 10.02 ± 1.6 | 9.3 ± 1.5 | 9.1 ± 1.2 | < 0.001 |
| MCV (fL) | 72.8 ± 5.4 | 71.2 ± 4.1 | 71.5 ± 4.7 | < 0.001 |
| MCH (pg) | 30.8 ± 7.4 | 30.2 ± 3.1 | 29.6 ± 3.3 | < 0.001 |
| MCHC (g/dL) | 32.07 ± 5.3 | 32.1 ± 3.6 | 32.6 ± 4.2 | < 0.001 |
| HCT (%) | 23.7 ± 3.4 | 23.7 ± 2.6 | 21.9 ± 2.0 | < 0.001 |
| HbF (%) | 25.1 ± 4.4 | 20.18 ± 3.7 | 13.1 ± 4.6 | < 0.001 |
Abbreviations: HbF, fetal hemoglobin; HbSS, hemoglobin sickle shaped; HCT, hematocrit; HMOX1, heme oxygenase-1; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; RBC, red blood cell.
| Hematological parameters | HMOX1(TT) n = 18(32%) | HMOX1(AT) n = 22(39%) | HMOX1(AA) n = 15(28%) | p-Value |
|---|---|---|---|---|
| RBC (millions of cells/µL) | 3.5 ± 1.7 | 3.3 ± 1.7 | 2.8 ± 1.5 | 0.001 |
| Hb (g/dL) | 10.07 ± 1.3 | 9.3 ± 1.6 | 9.1 ± 1.4 | < 0.001 |
| MCV (fL) | 72.8 ± 6.3 | 71.2 ± 4.7 | 68.5 ± 6.5 | < 0.001 |
| MCH (pg) | 34.8 ± 5.4 | 32.6 ± 4.5 | 32.6 ± 3.5 | < 0.001 |
| MCHC (g/dL) | 33.07 ± 3.2 | 30.7 ± 4.2 | 30.6 ± 3.2 | < 0.001 |
| HCT (%) | 23.7 ± 3.4 | 23.7 ± 2.6 | 21.9 ± 2.0 | < 0.001 |
| HbF (%) | 36.1 ± 4.7 | 32.1 ± 5.2 | 17.4 ± 4.7 | < 0.001 |
Abbreviations: HbF, fetal hemoglobin; HbSβ, sickle-beta thalassemia; HCT, hematocrit; HMOX1, heme oxygenase-1; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; RBC, red blood cell.
| HMOX1 polymorphism genotype and allele | HbSS, n = 65 | Controls n = 50 | p-Value, chi-squared |
|---|---|---|---|
| AA | 16 (25%) | 18(36%) | 0.1395,2.183 |
| AT | 29 (45%) | 25 (50%) | 0.5050, 0.444 |
| TT | 20(30%) | 7 (14%) | 0.0072, 7.212 |
| A | 47.5% | 61% | |
| T | 52.5% | 39% |
Abbreviations: HbSS, hemoglobin sickle shaped; HMOX1, heme oxygenase-1.
| HMOX1 polymorphism genotype and allele | Sβ, n = 55 | Controls, n = 50 | p-Value, chi-squared |
|---|---|---|---|
| AA | 15 (28%) | 18 (36%) | 0.4090,0.682 |
| AT | 22 (40%) | 25 (50%) | 0.2786, 1.174 |
| TT | 18(32%) | 7 (14%) | 0.0150, 0.745 |
| A | 48% | 59.5% | |
| T | 52% | 40.5% |
Abbreviations: HbSβ, sickle-beta thalassemia; HMOX1, heme oxygenase-1.
Discussion
The phenotypic–genotypic diversity and genetic heterogeneity of SCD along with multifactorial influence complicates the assessment of prognostication in these patients. Various factors implicated in modification of this disorder include HbF concentration, β-globin gene cluster haplotype that will ultimately influence its clinical severity.[18] Intracellular distribution of HbF is a strong modifier of SCD and studies have shown that elevated HbF levels tend to reduce the severity of SCD by reducing the sickling and its manifestations.[19] Higher levels of HbF (mean 22.3 ± 6.9%; range: 3.9–40.9%) are found in Indian SCD patients.[20] The βS gene is linked to an Asian haplotype and the XmnI restriction site, but these factors alone cannot explain the high HbF concentration.[21] Limited data is available regarding the co-expression of HbF modifier like HMOX1 with SCD and its influence on the disease severity and phenotype. To best of our knowledge, this is the first study in HbSS and HbSβ patients to investigate the prevalence and impact of HMOX1-polymorphisms on HbF levels and disease severity in Indian population.
HO enzyme encoded by HMOX1 gene located in long arm of chromosome 22 plays pivotal role common as an antioxidant enzyme in neutralizing the toxic free heme into three biologically active by products, that is, biliverdin, free iron, and carbon monoxide and plays a vital role in the protection of cells and DNA from reactive oxygen species damage. HMOX1 polymorphism rs2071746: A > T has been studied in various diseases to evaluate its role in ischemia and hypertension.[22] However, there is sparse data on the association of this polymorphism with SCD as a result of its impact on HbF. βs globin gene haplotypes are evaluated to assess the severity of disease. Some studies have shown that SNPs in some genes in HBBP1 and out of HMOX1 β-globin gene cluster can affect disease severity.[23,24]
A previous study from Egyptian population in year 2019 reported the prevalence of HMOX1 polymorphic AT and TT genotypes in patient and control group as 92 and 85% respectively.[25] Furthermore, a study in the Brazilian population in year 2020 showed that the HMOX1 rs2071746 genotype frequencies were 24.3% (AA), 48.6% (AT), and 27.0% (TT) in the SCA patient group and 28.2% (AA), 52.7% (AT), 19.1% (TT) in the control group.[26]
In this study, 29/65 (44.61%) heterozygous, 20/65 (30%) homozygous, and 16/65 (24.61%) were found wild genotype. However, AT was the most common genotype in HbSS patients and healthy individuals with 44.61 and 50% prevalence, respectively. This was concordance with Jaseb et al.[27] Out of 55 HbSβ, 22 cases (40%) were heterozygous, 18 (32%) were homozygous, and 15 (28%) cases were wild-type and this is in contrast to Bakr et al 2019, who reported a very low frequency of wild genotype (6.1%) and a much higher frequency of mutant genotype (93.9%) in Egyptian patients.[26] Furthermore, our study results suggest that SCD patients harboring mutant HMOX1 had less frequent vaso-occlusive crises, less incidence of blood transfusion, stroke, and hospitalization with improved hematological as well as clinical parameters. This is also in accordance with the data reported by Bakr et al.[26]
HMOX1 can ameliorate vaso-occlusion and prevent vascular inflammation via converting damaging heme into biliverdin and carbon monoxide molecules that have antioxidant, anti-inflammatory properties. Studies have shown that the T allele of rs2071746: A > T polymorphism leads to decreased expression of HMOX1 gene. There are two hypotheses regarding influence of this polymorphism on HbF levels; first this polymorphism in promoter region causes decreased production of HO protein, which increases the concentration of free heme in circulation. This leads to slow erythropoiesis and reduced production of erythroblasts which result in an augmented production of HbF, and second the homozygous mutant TT genotype may indirectly affect the genes associated with it, causing increased HbF production.[28–30]
Our findings showed the higher levels of HbF in SCD patients with HMOX1 gene polymorphism that was in line with the results of Bhagat et al and Keikhaei et al.[31,32] In our study population, TT genotype in HMOX1 gene (rs2071746: A > T polymorphism) had a significant correlation in relation to increasing HbF level in SCD (p < 0.001). This was in line with Gil et al 2013 and Jaseb et al, who found association of this genotype with increased levels of HbF (p = 0.0131, p < 0.019). However, the mean steady state HbF concentrations in our study was higher in the all three genotypes when compared to Bakr et al[26] and Gil et al[13] possibly due to the Arab-Indian haplotype of βS gene that is associated with higher HbF levels.
In conclusion, we found significant association between the TT genotype of the rs2071746:A > T polymorphism of HMOX1 gene, and increased levels of HbF. We suggest this polymorphism can be considered as novel marker related to HbF levels in Indian SCD patients so that it can serve as a new prognostic marker for guiding the clinicians to formulate management protocol among Indian SCD patients. However, further studies in larger subset of patients are suggested to strengthen our findings.
Authors' Contributions
H.R.P. designed the study, performed experimental studies, and drafted the manuscript. K.S. compiled the data and contributed to writing. R.R. and J.D. performed analysis and helped in drafting the manuscript. S.T. reviewed the manuscript and helped in shaping the manuscript. T.S., R.S., and M.M. provided valuable suggestions and clinical outputs. All authors read and approved the final the manuscript.
Statement of Informed Consent
All persons gave their informed consent prior to their inclusion in the study.
Conflict of Interest
None declared.
Funding
Authors would like to acknowledge the financial support given by Indian Council of Medical Research (ICMR)-56/1/2019-HAE/BMS-), New Delhi, India, for the present study.
References
- Clinical aspects of sickle cell anaemia in adults and children. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, eds. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (1st). Cambridge, England: Cambridge University Press; 2001. p. :611-670.
- [Google Scholar]
- Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762-769.
- [CrossRef] [PubMed] [Google Scholar]
- Geographical survey of β S-globin gene haplotypes: evidence for an independent Asian origin of the sickle-cell mutation. Am J Hum Genet. 1986;39(02):239-244.
- [Google Scholar]
- Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644.
- [CrossRef] [PubMed] [Google Scholar]
- Sickle cell disease pathophysiology. In: Higgs DR, Weatherall DJ, eds. Bailliere's Clinical Haematology: The Haemoglobinopathies. Vol 6. London: Bailliere Tindall; 1993. p. :57-91.
- [CrossRef] [PubMed] [Google Scholar]
- Beta thalassaemia. In: Rodgers GP, ed. Bailliere's Clinical Haematology, Sickle Cell Disease and Thalassaemia. Vol 11. London: Bailliere Tindall; 1998. p. :91-126.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000a;95(01):342-346.
- [CrossRef] [PubMed] [Google Scholar]
- A candidate gene study of F cell levels in sibling pairs using a joint linkage and association analysis. GeneScreen. 2000b;1:9-14.
- [CrossRef] [Google Scholar]
- Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87(08):795-803.
- [CrossRef] [PubMed] [Google Scholar]
- Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood. 2012;120(09):1961-1962.
- [CrossRef] [PubMed] [Google Scholar]
- Sickle cell disease in Orissa State, India. Lancet. 1986;2(8517):1198-1201.
- [CrossRef] [PubMed] [Google Scholar]
- High frequency of the CCR5delta32 variant among individuals from an admixed Brazilian population with sickle cell anemia. Braz J Med Biol Res. 2003;36(01):71-75.
- [CrossRef] [PubMed] [Google Scholar]
- Polymorphism in the HMOX1 gene is associated with high levels of fetal hemoglobin in Brazilian patients with sickle cell anemia. Hemoglobin. 2013;37(04):315-324.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br J Haematol. 2017;178(04):629-639.
- [CrossRef] [PubMed] [Google Scholar]
- Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64(01):72-80.
- [CrossRef] [PubMed] [Google Scholar]
- Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2(10):2557-2568.
- [CrossRef] [PubMed] [Google Scholar]
- Heme degradation and vascular injury. Antioxid Redox Signal. 2010;12(02):233-248.
- [CrossRef] [PubMed] [Google Scholar]
- Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129(04):465-481.
- [CrossRef] [PubMed] [Google Scholar]
- DNA sequence variation associated with elevated fetal G gamma globin production. Blood. 1985;66(04):783-787.
- [CrossRef] [PubMed] [Google Scholar]
- Higher fetal hemoglobin concentration in patients with sickle cell disease in eastern India reduces frequency of painful crisis. Eur J Haematol. 2009;83(04):383-384.
- [CrossRef] [PubMed] [Google Scholar]
- Fetal hemoglobin levels and β (s) globin haplotypes in an Indian populations with sickle cell disease. Blood. 1987;69(06):1742-1746.
- [CrossRef] [PubMed] [Google Scholar]
- A promoter variant of the heme oxygenase-1 gene may reduce the incidence of ischemic heart disease in Japanese. Atherosclerosis. 2004;173(02):315-319.
- [CrossRef] [PubMed] [Google Scholar]
- Influence of BCL11A, HBS1L-MYB, HBBP1 single nucleotide polymorphisms and the HBG2 XmnI polymorphism On Hb F levels. Hemoglobin. 2012;36(06):592-599.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of Swiss-type heterocellular HPFH from XmnI-Gγ and HBBP1 polymorphisms on HbF, HbE, MCV and MCH levels in Thai HbE carriers. Int J Hematol. 2014;99(03):338-344.
- [CrossRef] [PubMed] [Google Scholar]
- β-globin gene cluster haplotypes in a cohort of 221 children with sickle cell anemia or Sβ0-thalassemia and their association with clinical and hematological features. Acta Haematol. 2010;124(03):162-170.
- [CrossRef] [PubMed] [Google Scholar]
- Implication of HMOX1 and CCR5 genotypes on clinical phenotype of Egyptian patients with sickle cell anemia. Ann Hematol. 2019;98(08):1805-1812.
- [CrossRef] [PubMed] [Google Scholar]
- Polymorphisms in the heme oxygenase-1 and bone morphogenetic protein receptor type 1b genes and estimated glomerular filtration rate in Brazilian sickle cell anemia patients. Hematol Transfus Cell Ther. 2020;16:30035-30043.
- [CrossRef] [PubMed] [Google Scholar]
- Association between beta globin haplotypes, HBBP1 and HMOX1 polymorphisms in relation to HbF among sickle cell anaemia patients: a study in Southwest Iran. Comp Clin Pathol. 2017;26:1149-1155.
- [CrossRef] [Google Scholar]
- HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum. 2007;56(12):3953-3958.
- [CrossRef] [PubMed] [Google Scholar]
- Heme oxygenase-1 deletion affects stress erythropoiesis. PLoS One. 2011;6(05):e20634.
- [CrossRef] [PubMed] [Google Scholar]
- Fetal haemoglobin and β-globin gene cluster haplotypes among sickle cell patients in Chhattisgarh. J Clin Diagn Res. 2013;7(02):269-272.
- [CrossRef] [PubMed] [Google Scholar]
- Beta-globin gene cluster haplotypes in Iranian sickle cell patients: relation to some hematologic. Iran J Blood Cancer. 2012;4(03):105-110.
- [Google Scholar]



