
Translate this page into:
Thanatophoric Dysplasia Type I: A Rare Case Report at Fetal Autopsy
Address for correspondence: Dr. Reshma S Davanageri, E-mail: reshdav@yahoo.co.in
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Thanatophoric dysplasia type 1 is a lethal congenital anomaly with skeletal dysplasia. It is characterized by short limb dwarfism, enlarged head with frontal bossing, short neck, protuberant abdomen. It is detected in early gestational period by ultrasonography. The other associated anatomical abnormalities and characteristic laboratory findings aid in the early diagnosis and further work-up.
Keywords
Fetal autopsy
short limb
thanatophoric dysplasia

INTRODUCTION
Thanatophoric dysplasia (TD) type 1 is a form of lethal osteochondrodysplasia which occurs sporadically and as a result of new autosomal dominant mutation.[1] The reported incidence is about 1 in 60,000 births.[2] Affected neonate shows marked underdevelopment of the skeleton and short limb dwarfism.[3] The anomaly results due to mutations of fibroblast growth factor receptor 3 gene (FGFR3) leading to the introduction of a new cysteine residue in various locations of the extramembranous segment of the receptor.[1] Diagnosis of this condition is made by ultrasonography usually in the second trimester.[3] We report one such rare case encountered at fetal autopsy at 20 weeks gestational age.
CASE REPORT
A male fetus of 20 weeks gestational age weighing 400 g was received at autopsy. The fetus was stillborn to a 26-year-old female with non-consanguineous marriage whose pregnancy was terminated after an ultrasonography, which showed micromelia. There was a history of termination of previous two pregnancies also. The first one was for congenital anomaly with short limbs and the second one for preeclampsia, with the fetus having normal limbs. The details regarding any other associated anomalies in the first pregnancy are not known. On examination, the fetus had skeletal dwarfism, with both the upper and lower limbs being shortened. Head was enlarged with bulging of forehead, eyes looked prominent and were widely spaced, nasal bridge was depressed and the neck was shortened. There was relative narrowing of the thorax with distended abdomen. Furthermore, abduction of bilateral hip joint was seen [Figure 1].
Laboratory investigations
With the above features, considering the possibility of TD type 1, an infantogram was done which assisted in making a diagnosis of this rare congenital anomaly. The findings on infantogram were hypoplastic lungs, modeling deformity with short and stubby iliac blades, femur, tibia, fibula, humerus, radius and ulna. Metatarsal bones were also short and stubby. Typical features such as telephone receiver like curved femora and curved clavicles resembling bicycle handle bars were noted. The ribs were short and horizontally placed [Figure 2]. To further support the diagnosis, histopathological examination of the sections from the knee joint revealed disorganized physeal growth zone with disordered proliferation and hypertrophy of chondrocytes and peripheral band of horizontally oriented fibrosis [Figure 3]. Taking into consideration the various anatomical abnormalities, radiological and histopathological findings, a final diagnosis of TD type 1 was made.
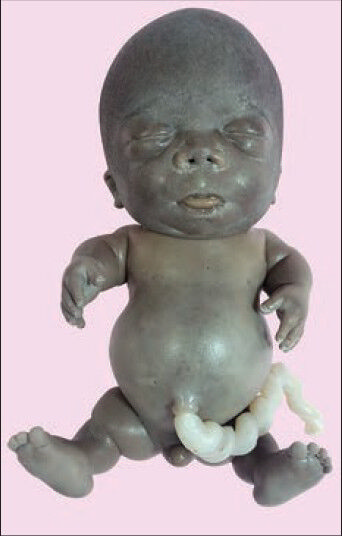
- Neonate with a large head, frontal bossing, prominent eyes, hypertelorism, depressed nasal bridge, short neck, narrow chest, protuberant abdomen, micromelia, bowing of legs, abduction of the hip joint
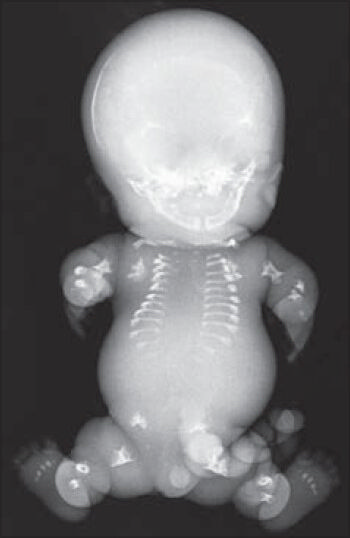
- Radiograph showing short ilia, short and curved femora, curved clavicle, short and horizontally placed ribs
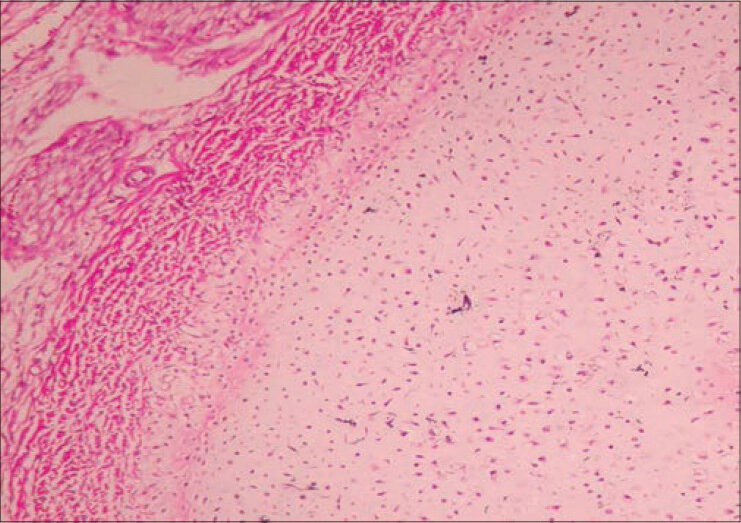
- Photomicrograph showing disorganization of physeal growth zone and peripheral band of horizontally oriented fibrosis
DISCUSSION
TD type 1 is a lethal skeletal dysplasia.[3] The name is derived from the Greek word meaning “death-bearing.”[4] This anomaly is detected early in gestation by ultrasonography.[1] The characteristic features include micromelia (short limbs), bowing of limbs, narrow thorax, protuberant abdomen.[5] Other features include polyhydromnios, large head with frontal bossing, depressed nasal bridge, prominent eyes, hypertelorism, short neck and small pelvis.[36] The classical features on radiography include severe platyspondyly, short ribs, small ilia, markedly shortened limb bones with telephone receiver-like curved femora and curved clavicles resembling bicycle handle bars. Histologically, nonspecific disturbance of enchondral ossification is seen, which is characterized by retardation of growth zone with disordered proliferative, hypertrophic chondrocytes. A band of horizontally oriented fibrosis is frequently seen at the periphery of the physis.[1]
TD type 1 differs from its reminiscent TD type 2 by few features. The latter shows cloverleaf skull with strikingly trilobed configuration and femora that are straight. TD type l may show cloverleaf skull, but it never reaches the strikingly trilobed configuration. Histologically, TD type 2 shows the presence of ossified cartilage canals in the physes of appendicular bones.[1]
TD type 1 is caused by mutation in FGFR3 located on chromosome 4 which results in activation of FGFR3 tyrosine kinase independently of ligands such as fibroblast growth factor 8, leading to decreased apoptosis and increased proliferation.[6] The affected neonates are usually stillborn or die shortly after birth from respiratory failure. However a small number of individuals have survived into childhood and a very few beyond, requiring respiratory support.[4]
The 3D anatomy scan and molecular confirmation are helpful in early diagnosis and genetic counseling of TD. Since the TD usually occurs sporadically, the recurrence risk is low, with estimated rate of only 2%.[3] However in our case, there could have been a recurrence, with autosomal dominant mode of inheritance, which is less common.
The present case showed all the classical abnormalities of TD type 1 on gross, radiological and histopathological examination. This case is reported for the rare incidence and probable mode of occurrence and to review in detail the various anatomical abnormalities and laboratory findings that are encountered in this anomaly which would aid in the early diagnosis and further work up.
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- Skeletal system. In: Enid GB, ed. Potter's Pathology of the Foetus, Infant and Child (2nd ed). Philadelphia: Mosby Elsevier; 2007.
- [Google Scholar]
- The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: The achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev. 2000;21:23-39.
- [Google Scholar]
- Long-term survival in typical thanatophoric dysplasia type 1. Am J Med Genet. 1997;70:427-36.
- [Google Scholar]
- Sonographic diagnosis of thanatophoric dwarfism in utero. J Ultrasound Med. 1982;1:337-9.
- [Google Scholar]
- Brain and bone abnormalities of thanatophoric dwarfism. AJR Am J Roentgenol. 2009;192:48-51.
- [Google Scholar]



