
Translate this page into:
Morquio Syndrome Presenting with Dural Band Pathology: A Case Report

Address for correspondence Dr. Arulselvi Subramanian, MD, Department of Lab Medicine, Jai Prakash Narayan Apex Trauma Center, AIIMS, New Delhi 110029, India (e-mail: arulselvi.jpnatc@gmail.com).
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Morquio syndrome is caused by the deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS) enzyme, which is required for the catabolism of glycosaminoglycans (namely, chondroitin-6-sulfate and keratan sulfate). Pathogenic accumulation of these glycosaminoglycans occurs throughout the body. The various organs and tissues affected are bones, cartilage, tendon, teeth, trachea and lungs, heart, cornea, skin and connective tissues.
Here, we present a case of Morquio syndrome. A 16-year-old boy presented with multiple skeletal abnormalities, including cervicomedullary compression by dorsal dural band in foramen magnum. The dural band was resected during the surgery to relieve compression and sent for histopathological examination. This case report not only reviews the clinical features and shows rare dural band histopathological findings but also mentions a note on the future therapies of this syndrome.
Keywords
cervicomedullary compression
dural calcification
Morquio syndrome

Introduction
Morquio syndrome, also known as mucopolysaccharidosis IV (MPS IV), is a lysosomal storage disease (LSD), which is caused by the absence of N-acetylgalactosaime-6-sulfate sulfatase (GALNS). Deficiency of GALNS causes abnormal accumulation of specific glycosaminoglycans (GAGs) like keratan sulfate (KS) and chondroitin-6-sulfate (C6S), mainly in the cartilage and its extracellular matrix (ECM), resulting in incomplete and distorted endochondral ossification and leading to successive systemic skeletal dysplasia.1-7 MPS are usually inherited in an autosomal recessive manner.
The exact incidence of Morquio syndrome worldwide is unknown but is estimated to be between 1 in 75000 population in Northern Ireland, 1 in 200,000 population in British Columbia, and 1 in 640,000 live births in Western Australia.8 In India, MPS accounts for 22% of all the LSD. MPS IV is the most common, comprising 26% out of a total of 85 MPS cases identified in India in a study conducted by Khan et al in 2017.9
At birth, patient with Morquio syndrome appears healthy and has normal intelligence. However, later on presents with multiple anomalies like disproportionate dwarfism, anterior beaking of lumbar spine, pectus carinatum, flaring of the rib cage, epiphyseal dysplasia of joints, knock-knee, laxity of joints, corneal opacities, dental abnormalities, and cardiopulmonary complications.1-7,10 Many patients become physically disabled in their second decade and undergo multiple orthopedic surgeries to lessen serious medical complications.5-7 The common ocular findings reported in Morquio syndrome are progressive corneal clouding, corneal opacification, retinopathy, optic nerve swelling and atrophy, ocular hypertension and glaucoma, astigmatism, and punctiform lens opacities.11,12 MPS I, II, VI (but not MPS III, IV) involving derangement of dermatan sulfate catabolism exhibit severe cardiovascular manifestations such as commonly valvular disease and, rarely, dilated cardiomyopathy and coronary involvement.13,14 The main causes of mortality and morbidity are valvular heart disease, respiratory failure by way of obstructive and restrictive lung disease and spinal cord injury due to spinal cord compression and instability of vertebral joints5-7,15
Diagnosis is typically based on clinical examination, radiographs, histopathology, urinary GAG tests and enzymatic activity of GALNS in blood and fibroblasts.5 Following the diagnosis, MPS IV requires an interdisciplinary approach toward patient care. Despite the devastating skeletal disease, there has been only a few studies reporting histological evaluations of bone and cartilage pathology in MPS IV,16,17 as Morquio syndrome is a rare entity and there is a paucity of cases reporting the dural band pathology in the literature.
Case History
Here, we present a case of a 16-year-old male, a known case of Morquio syndrome, who exhibited normal milestones till the age of 6 years after which vertebral abnormality started becoming apparent. The physical appearance was characterized by short stature, short neck, protuberant chest, scoliosis, and facial features presented with frontal bossing and flat nasal bridge. He has normal intelligence. There was no family history. His parents did not belong to different ethnicity and they had a nonconsanguineous marriage.
The patient had muscular weakness, which led to falls sometimes while walking and progressive spastic quadriparesis for 10 years. He now presented with cervicomedullary compression by dorsal dural ring in foramen magnum for which he underwent corpectomy with autologous fibular graft placement and anterior cervical plating. The orthopedic and radiographic evaluation indicated kyphotic neck with thoracic deformity. MRI of whole spine showed absent to severe hypoplastic odontoid process of C2 along with severe stenosis from foramen magnum to C2 level and severe compression of cord. Cervicodorsolumbar bullet/hook-shaped vertebral bodies with cord changes were seen at these levels (►Fig. 1). There were no ocular or cardiac manifestations in our patient. A diagnosis of MPS type IV was reached based on the clinical, radiographic and histopathological findings of the patient. Dural band removed from surgery was sent for histopathological examination, which showed severe vacuolation and ballooning of chondrocytes along with fibrocollagenous and fibrocartilaginous tissue. Impression given by analyzing H & E and PAS-stained slides was dural band with changes due to Morquio syndrome. (►Figs. 23-4) The diagnosis would need to be confirmed by analysis of GALNS enzyme activity and molecular genetic detection of GALNS gene. Unfortunately, parents’ consent could not be obtained for this analysis.
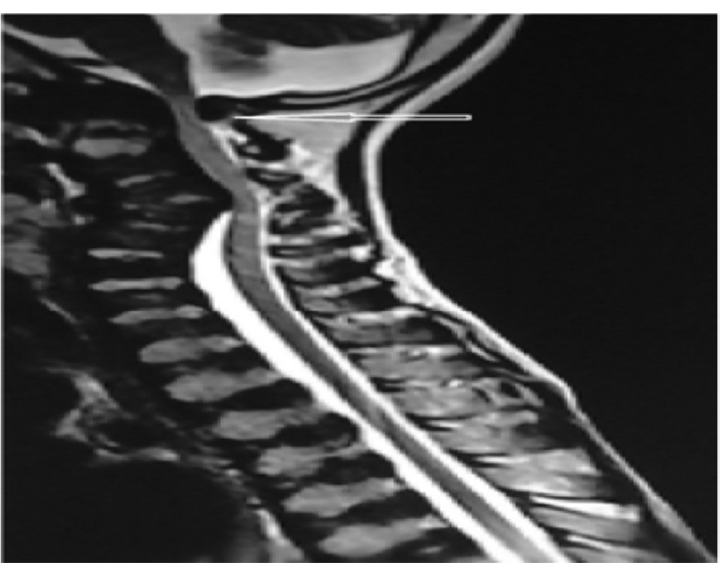
- MRI whole spine- craniovertebral junction presenting compression at C1 level (arrow).

- H&E stain; 4× poorly organized fibrocollagenous and fibrocartilaginous tissue in the dural band.

- PAS stain; 10× vacuolated and ballooned chondrocytes in fibrocartilaginous tissue.

- Masson trichrome stain; 40× chondrocytes are heterogeneously distributed, oversized, severely vacuolated and ballooned.
Discussion
MPS type IV (osteochondrodystrophia) is a rare multisystem disorder which presents with several musculoskeletal defects and exhibits a chronic progressive course. The clinical condition was first described independently in 1929 by Morquio and Brailsford. Matalon et al in 1974 identified the deficiency of enzyme GALNS, causing intracellular accumulation of keratan sulfate and chondroitin-6-sulfate responsible for Morquio syndrome type IVA.18 MPS type IVB is due to deficiency of β-galactosidase which accumulates keratan sulfate.19 Their chromosomal locations are 16q24.3 for GALNS and 3p21.33 for β-galactosidase. A nonkeratan sulfate excretion variety is labeled as MPS type IVC.20
The clinical presentation of Morquio syndrome is slightly complex and is hard to diagnose in the initial few years of life. The children affected with Morquio syndrome have normal appearance during the first few years of life, thus making the condition less recognizable. However, Maccari et al stated that urinary uronic acid GAG levels are very high in the neonatal period. They used HPLC MS and tandem MS for proving these GAGs as a biomarker and a screening tool for MPS in a newborn of age 2 to 3 days.21 The GAG aggregation in the body may cause premature death, with a life expectancy ranging from 10 to 20 years of age to almost normal in some patients.6 A family tendency of unknown etiology has also been reported by Rekha et al, where three siblings in the same family were affected with the syndrome; 22 however, in our case, no such familial tendency was noted.
Khan et al stated that for the degradation of GAGs, lysosomal enzymes are critical to provide a normal function of tissues and ECM. As many as 328 mutations (missense/nonsense mutations [commonest], deletions, splice site mutations, insertions) in GALNS gene are responsible for the lack of lysosomal enzymes, resulting in wide phenotypic variability.9 The insufficient amount of lysosomal hydrolases result in aggregation of nondegraded GAGs, triggering release of numerous mediators including growth factors and cytokines, and resulting in chronic inflammation (T cell-mediated)17 like arthritis, valvular heart disease as well as degenerative changes in chondrocytes, abnormal ECM and collagen. These damage the entire process of ossification and also cause laxity of joints due to high-expression of collagen type I and low-expression of collagen type II, resulting in reduced movement in knee and ankle joints.16
Also, these GAGs are secreted into the blood circulation and then excreted in urine, causing marked elevation of blood and urine KS levels, which is compatible with a severe clinical phenotype of the patient, indicating that KS is a good biomarker, especially at a progressive stage.
Our case highlights the dural band pathology in Morquio syndrome, showing severe ballooning and vacuolation of chondrocytes. Kalteis described a case of Morquio syndrome of a 36-year-old female patient in whom severely vacuolated chondrocytes were seen in both knee joints.23 Yasuda et al showed the appearance of foam cells and macrophages in lung, aorta, heart valves, heart muscle, trachea, visceral organs, and bone marrow in an autopsied case.24
Doherty et al reported a case of an autopsied 23-year-old male with MPS IVA. Histopathological examination of postmortem tissues revealed severely ballooned and vacuolated chondrocytes in trachea, humerus, knee cartilage, and lung bronchus along with systemic storage materials in various other tissues.25 De Franceschi et al revealed the histopathological findings of articular cartilage taken from femoral condyle of two siblings with Morquio syndrome. The features showed an unorganized tissue structure, with cells larger than normal chondrocytes and with heterogeneous distribution.16
Morquio syndrome needs to be differentiated from other MPS, based on the presentation. Unlike other MPS, patients with Morquio syndrome have normal intellectual abilities, less coarse facial features, atlantoaxial instability, wrist weakness, and knock-knee MPS type I, II, VII showing soft-tissue storage and skeletal disease with or without brain disease. MPS type VI predominantly shows soft tissue and skeletal involvement. Primarily, central nervous system (CNS) involvement is observed in type III MPS. MPS IVA and IVB (β-galactosidase enzyme deficiency causes only KS accumulation) share several common disease characteristics, but MPS IVA is more dreadful than MPS IVB.
Contrary to MPS IV, GM1 gangliosidosis patients have CNS impairment, developmental delay, craniofacial dysmorphia and fetal hydrops. Radiographically and clinically, spondyloepiphyseal dysplasia congenita (SEDc) is similar to MPS IVA, but SEDc patients have joint stiffness as opposed to hypermobile joints of MPS IV. Distinct to MPS IVA, Legg–Calve–Perthes disease (COL2A1 mutations) affects only the hip. In contrast to spine involvement of Morquio syndrome, juvenile idiopathic arthritis and rickets show joint pain, deformities and widening of joints without spine involvement.12,26
Early diagnosis indicates early intervention and specific therapies for Morquio syndrome. The future therapies for MPS include enzyme replacement therapy, gene therapy, substrate replacement therapy and hematopoietic stem cell transplantation. Genetic counselling and prenatal diagnosis using amniotic fluid cells or chorionic villus biopsy should be conducted. Recently, ELISA has been used for quantification of Keratan sulfate in urine and blood in MPS type IVA patients.27 The current clinical under trial drug for Morquio A syndrome is BMN 110 which is an enzyme replacement of GALNS.27
Conclusion
Dural band pathology was an unusual finding in this rare disorder. Reporting of this case is important in view of paucity of epidemiological data regarding exact incidence of this rare syndrome. It is also essential to assess the risk of this syndrome in the other members of the affected families as well as for genetic counselling. Early diagnosis indicates prompt intervention and specific therapies and also lowers the number of newborns with LSDs. Also, an increase in the life expectancy can be anticipated on starting these treatments at early stages of the disease.
Conflicts of Interest
None declared.
Reference
- The mucopolysaccharidoses. In: Scriver C.R.F, Beaudet A.L. W.S., Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease (8th). New York: McGraw-Hill; 2001. p. :3421-3452.
- [Google Scholar]
- Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns-/-) produced by targeted disruption of the gene defective in Morquio A disease. Hum Mol Genet. 2003;12(24):3349-3358.
- [CrossRef] [PubMed] [Google Scholar]
- Development of MPS IVA mouse (Galnstm(hC79S.mC76S)slu) tolerant to human N-acetylgalactosamine-6-sulfate sulfatase. Hum Mol Genet. 2005;14(22):3321-3335.
- [CrossRef] [PubMed] [Google Scholar]
- Murine model (Galns(tm(C76S)slu)) of MPS IVA with missense mutation at the active site cysteine conserved among sulfatase proteins. Mol Genet Metab. 2007;91(03):251-258.
- [CrossRef] [PubMed] [Google Scholar]
- International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30(02):165-174.
- [CrossRef] [PubMed] [Google Scholar]
- Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12(06):931-945.
- [CrossRef] [PubMed] [Google Scholar]
- Current and emerging treatments and surgical interventions for Morquio A syndrome: a review. Res Rep Endocr Disord. 2012;2012(02):65-77.
- [CrossRef] [PubMed] [Google Scholar]
- Morquios syndrome: A rare case report. J Indian Acad Oral Med Radiol. 2013;25:63-65.
- [CrossRef] [Google Scholar]
- Epidemiology of mucopolysaccharidoses. Mol Genet Metab. 2017;121(03):227-240.
- [CrossRef] [PubMed] [Google Scholar]
- The Morquio syndrome (mucopolysaccharidosis IV): Morphologic and biochemical studies. Johns Hopkins Med J. 1975;137(04):176-183.
- [Google Scholar]
- Manifestations ophtalmologiques de la maladie de Morquio. [Ocular manifestations in patients affected by Morquio syndrome (MPS IV)] J Fr Ophtalmol. 2010;33(09):617-622.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular genetics and metabolism, special edition: diagnosis and prognosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2018;125(1-2):18-37.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(06):1183-1197.
- [CrossRef] [PubMed] [Google Scholar]
- Early diagnosis and management of cardiac manifestations in mucopolysaccharidoses: a practical guide for paediatric and adult cardiologists. Ital J Pediatr. 2018;44(Suppl. 02):122.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013;36(02):309-322.
- [CrossRef] [PubMed] [Google Scholar]
- A molecular and histological characterization of cartilage from patients with Morquio syndrome. Osteoarthritis Cartilage. 2007;15(11):1311-1317.
- [CrossRef] [PubMed] [Google Scholar]
- Deficiency in N-acetylgalactosamine-6-sulfate sulfatase results in collagen perturbations in cartilage of Morquio syndrome A patients. Mol Genet Metab. 2009;97(03):196-201.
- [CrossRef] [PubMed] [Google Scholar]
- Deficiency of chondroitin sulfate N-acetylgalactosamine 4-sulfate sulfatase in Maroteaux-Lamy syndrome. Biochem Biophys Res Commun. 1974;61(04):1450-1457.
- [CrossRef] [PubMed] [Google Scholar]
- Spondyloepiphyseal dysplasia, corneal clouding, normal intelligence and acid beta-galactosidase deficiency. Clin Genet. 1976;9(05):495-504.
- [CrossRef] [PubMed] [Google Scholar]
- Total and single species of uronic acid-bearing glycosaminoglycans in urine of newborns of 2-3days of age for early diagnosis application. Clin Chim Acta. 2016;463:67-72.
- [CrossRef] [PubMed] [Google Scholar]
- Mucopolysaccharidoses type IV A (Morquio syndrome): a case series of three siblings. J Indian Soc Pedod Prev Dent. 2012;30(01):66-69.
- [CrossRef] [PubMed] [Google Scholar]
- Arthroscopic and histologic findings in Morquio’s syndrome. Arthroscopy. 2005;21(02):233-237.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenesis of Morquio A syndrome: an autopsied case reveals systemic storage disorder. Mol Genet Metab. 2013;109(03):301-311.
- [CrossRef] [PubMed] [Google Scholar]
- Natural history of Morquio A patient with tracheal obstruction from birth to death. Mol Genet Metab Rep. 2017;14:59-67.
- [CrossRef] [PubMed] [Google Scholar]
- Mucopolysaccharidosis type IVA (Morquio A): a close differential diagnosis of spondylo-epiphyseal dysplasia. BMJ Case Rep. 2017;2017 bcr-2017-221156
- [CrossRef] [PubMed] [Google Scholar]
- A rare case of mucopolysaccharidosis. Indian J Clin Biochem. 2014;29(01):101-106.
- [CrossRef] [PubMed] [Google Scholar]



