
Translate this page into:
X-linked Hyper-IgM Syndrome with Bronchiectasis
Address for correspondence: Dr. Devki Nandan, E-mail: devkinandan2002@yahoo.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The X-linked hyper-immunoglobulin M syndrome (HIGM-1) is a rare genetic disorder characterized by elevated serum IgM levels and low to undetectable levels of serum IgG, IgA and IgE. These patients characteristically present with recurrent sinopulmonary infections and recurrent diarrhea. They also have high susceptibility for Pneumocystis jiroveci (PJ) pneumonia. Herein, we report a case of HGM-1 in a 5-year-old boy who presented with bronchiectasis and, possibly, PJ pneumonia. The diagnosis was established on the basis of clinical features, immune profile, whole blood flow cytometry and history of two male sibling's death due to recurrent pneumonia and diarrhea.
Keywords
Bronchiectasis
children
Pneumocystis jiroveci
X-linked hyper-IgM syndrome

INTRODUCTION
Hyper-immunoglobulin M syndrome (HIGM) is a rare heterogeneous group of disorders, 70% of which are due to CD40-ligand deficiency.[1] These patients are susceptible to recurrent sinopulmonary infections and opportunistic infections, especially Pneumocystis jiroveci pneumonia. Bronchiectasis is characterized by abnormal dilated thick-walled bronchi that are inflamed and colonized by bacteria. After excluding cystic fibrosis, tuberculosis and congenital lung abnormalities, the patient should be investigated for primary immunodeficiencies.[2]
On searching the literature for the youngest and only case of HIGM with bronchiectasis, we could found data of a 7-year-old girl published by Montella et al.[3] To the best of our knowledge, the case described here is the youngest case of HIGM-1 presenting with bronchiectasis and, possibly, PJ pneumonia.
CASE REPORT
A 5-year-old boy, born of consanguineous (4th degree) marriage, presented with complaints of productive cough and right ear discharge since 1 year, fever and respiratory distress for 25 days and oral thrush since 20 days.
He had a history of five admissions in various hospitals for pneumonia since the age of 1 year.
The first episode of pneumonia occurred at the age of 6 months. He also had a history of repeated diarrhea (six to seven episodes every year) since the age of 6 months. Antitubercular therapy was given on an empirical basis at the age of 2.5 years, without any significant improvement. Two of his elder male siblings died because of recurrent pneumonia and diarrhea at the age of 2 years and 8 years. Onset of recurrent pneumonia and diarrhea in both the siblings was at about 8 months of age.
On examination, his heart rate was 120/min, respiratory rate was 66/min, SpO2 was 91% (on room air) and grade 2 clubbing was present. There was no cyanosis, icterus and palpable lymph nodes. Oral thrush and postnasal drip was present. Tonsils were small. Right side maxillary area tenderness and right ear discharge (yellowish-green) were also noted. There was no mastoid tenderness. On anthropometry, he was wasted and stunted. The respiratory system examination revealed tachypnea, chest retractions and bilateral diffuse crepitations. Other organ systems were essentially normal.
His blood counts revealed total leukocyte count -27,000/cc (P75) and the platelet counts and erythrocyte sedimentation rate were normal. Arterial blood gas revealed pH -7.32, PCO2 -53.1 mmHg, PaO2 -60 mmHg and HCO3 -26.9 mEq/L. Liver function test, renal function tests and coagulation profile were normal.
The serum lactate dehydrogenase level (LDH) was raised. The oral swab culture revealed Candida albicans and urine microscopic examination showed fungal hyphae. Ear swab and bronchoalveolar lavage cultures were positive for Pseudomonas aeruginosa. On bronchoscopy, the right bronchial tree showed collapse of some sub-segmental bronchi and proximal bronchiectasis. Work up for other causes of bronchiectasis was negative [Table 1].

High serum IgM level and no detectable serum levels of IgG and IgA [Table 1] suggested HIGM syndrome. His whole blood flow cytometry revealed reduced expression of CD40L (CD154) on activated T lymphocytes after stimulation [Table 1] thus confirming the diagnosis of HIGM-1.
Chest roentgenogram performed 1 month before the admission and at the time of admission showed persistent opacity in the right middle zone. An ultrasound of the abdomen did not reveal any abnormality. High-resolution computed tomography (HRCT) of the chest demonstrated bronchiectatic changes in the right lower lobe, right middle lobe, left lower lobe and lingular lobe [Figure 1a and b].
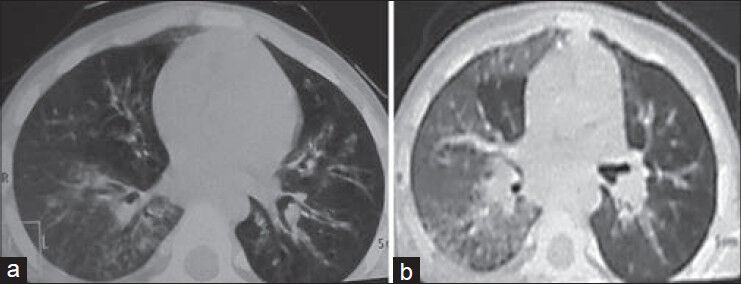
- Chest high-resolution computed tomography. (a) Areas of ground-glass haze and nodular air space lesions in a branching pattern (tree in bud) in the right lower lobe, dilated and thickened bronchi in the left lower lobe, lingular lobe and right middle lobe. (b) Ground-glass haze in the right lung with air space lesions in the right lower lobe
The patient presented with severe respiratory distress, for which he was put on mechanical ventilation. Intravenous (IV) ceftriaxone and cloxacillin were started in addition to nebulizations with 3% saline and chest physiotherapy. On the basis of culture sensitivity (Pseudomonas growth) report of the ear swab and bronchoalveolar lavage, antibiotics were upgraded to inj. Piperacillin-Tazobactum and Amikacin, and were continued for 14 days. IV liposomal Amphotericin B and Co-Trimoxazole was given in view of probable invasive candidiasis[4] and PJ pneumonia, respectively. IV immunoglobulin (0.6 gm/kg, single dose) was also given.
He did respond to the treatment well, and was discharged in satisfactory condition and is in regular follow-up. He is getting oral Co-Trimoxazole prophylaxis, IVIG therapy (600 mg/kg ever 4 weeks), Salbutamol and Budesonide inhalation with MDI and spacer and 3% saline nebulization followed by chest physiotherapy at home.
DISCUSSION
X-linked HIGM syndrome results due to defects in class switch recombination caused by mutations in the CD40 ligand and nuclear factor kB essential modifier (NEMO).[5] T cell functions are also affected in both CD40L and NEMO deficiency, which explains the associated opportunistic infections.[5] X-linked forms are due to CD 40 ligand and NEMO gene defects. NEMO gene defect was ruled out in our case due to the absence of ectodermal dysplasia, mycobacterial infection, osteopetrosis and lymphedema. HIGM-1 usually presents around 6-9 months of age (when maternal antibodies decrease below the protective level) with recurrent sinopulmonary bacterial infections, opportunistic infections and recurrent diarrhea. Neutropenia, thrombocytopenia and anemia are also seen.[6]
Singleton et al. reported recurrent pneumonia as the major preceding medical condition leading to bronchiectasis.[7] Lodha et al. reported that 10 (52.6%) cases of recurrent pneumonia were associated with bronchiectasis.[8] Management of bronchiectasis includes antibiotics, bronchodilators, inhaled steroids, mucolytics and chest physiotherapy, along with treatment of underlying disorder.[9] The goal of therapy is to mobilize secretions and to reduce the infectious and inflammatory load, thereby preventing progression of airway destruction.
PJ pneumonia is being reported as the presenting feature in about 40% of HIGM-1 patents.[10] Festic et al. reported elevated LDH in 29 of 30 patients with non-HIV PJ pneumonia.[11] The classic presentation of PCP is a bilateral interstitial pattern, which may be characterized as finely granular, reticular or ground-glass opacities.[12] In the described patient, there was markedly elevated serum LDH level with ground-glass haze on contrast-enhanced computed tomography of the chest [Figure 1a and b], suggesting interstitial pneumonitis and hypoxemia suggesting possibility of PJ pneumonia. However; we could not recover PJ trophozoites and cysts in the bronchoalveolar lavage, which may be explained by the fact that BAL (Bronchoalveolar lavage) could only be performed after initial stabilization of the patient and Co-Trimoxazole was started empirically on the basis of raised LDH level with pneumonia and hypoxemia.
Appropriate treatment of infections and regular IVIG therapy (every 3-4 weeks) is considered the mainstay of therapy. Boiled drinking water to prevent Cryptosporidium infection and Co-Trimoxazole prophylaxis to prevent PJ pneumonia should be given. The only curative treatment currently available for HIGM-1 is allogeneic hematopoietic cell transplantation (HCT). However, liver damage and bronchiectasis may limit the success of bone marrow transplantation.[11]
Early diagnosis and appropriate management of recurrent/persistent pneumonia in cases of HIGM-1syndrome may prevent development of bronchiectasis, a serious complication that limits the success of bone marrow transplantation, a definite cure of HIGM-1 syndrome.
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539-41.
- [Google Scholar]
- Non-CF bronchiectasis: Does knowing the aetiology lead to changes in management? Eur Respir J. 2005;26:8-14.
- [Google Scholar]
- Hyper IgM syndrome presenting as chronic suppurative lung disease. Ital J Pediatr. 2012;38:45.
- [Google Scholar]
- European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group; National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis. 2008;46:1813-21.
- [Google Scholar]
- Clinical consequences of defects in B-cell development. J Allergy Clin Immunol. 2010;125:778-87.
- [Google Scholar]
- Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105:1881-90.
- [Google Scholar]
- Bronchiectasis in Alaska Native children: Causes and clinical courses. Pediatr Pulmonol. 2000;29:182-7.
- [Google Scholar]
- Acute respiratory failure due to pneumocystis pneumonia in patients without human immunodeficiency virus infection: Outcome and associated features. Chest. 2005;128:573-9.
- [Google Scholar]
- Imaging features of of pneumocystis carinii pneumonia. Crit Rev Diagn Imaging. 1999;40:251-84.
- [Google Scholar]



