
Translate this page into:
Prevalence of hemoglobin variants and hemoglobinopathies using cation-exchange high-performance liquid chromatography in central reference laboratory of India: A report of 65779 cases
Address for correspondence: Dr. Sandeep Warghade, Head of Department, Department of Hematology, Metropolis Healthcare Limited, Kurla (West), Mumbai, Maharashtra, India. E-mail: sandeep.warghade@metropolisindia.com
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
CONTEXT:
Hemoglobinopathies constitute the world's most common genetically inherited red blood cell disorder. Screening and accurate identification of hemoglobin (Hb) variants have become increasingly important in antenatal diagnosis and prevention of Hb disorders.
AIM:
The aim of this study was to screen and identify Hb fractions prevalent in the Central Reference Laboratory of India.
MATERIALS AND METHODS:
A total of 65,779 cases were screened for hemoglobinopathies on the bio-rad variant high-performance liquid chromatography (HPLC) system by beta-thalassemia short program. The retention times, proportion of the hemoglobin (%) and the peak characteristics for all hemoglobin fractions were recorded. Molecular analysis of the beta-globin gene was carried out by DNA sequencing on eight cases.
RESULTS:
Total number of abnormal Hb fractions on cation exchange-HPLC (CE-HPLC) was seen in 12,131 (18.44%) cases. Beta-thalassemia trait was the predominant genetic Hb disorder accounting for 7377 cases (11.21%) of the total cases. This was followed by sickle cell trait (2.01%), sickle cell disease (1.59%), beta-thalassemia syndrome (0.80%), HbE trait (0.79%), and borderline HbA2 (0.51%). Molecular characterization of eight rare cases of hemoglobin variants by beta-globin gene sequencing identified three cases of Hb Beth Israel, two cases of Hb Hofu trait, and one case each of Hb J Cambridge, Hb Mizunami, and Hb Sherwood Forest.
CONCLUSION:
Superior resolution, rapid assay time, and accurate quantification make CE-HPLC suitable for the routine investigation of hemoglobinopathies.
Keywords
Cation exchange-high-performance liquid chromatography
hemoglobin variants
hemoglobinopathies
India
thalassemia

Introduction
Hemoglobinopathies are the most common genetically inherited disorders. The figures of World Health Organization (WHO) estimate that approximately 5% of world's population are being carriers for the genetic hemoglobin (Hb) disorders. Every year, there are over 42 million carriers and more than 12,000 infants born with a major and clinically significant hemoglobinopathy. In India, the cumulative gene frequency of hemoglobinopathies is around 4.2%. Worldwide migration of human population, relatively higher frequency of consanguineous marriages in many of the high frequency countries, has equally contributed for the increased burden of hemoglobinopathies.[1]
Abnormalities of Hb involve thalassemias and Hb variants which are due to abnormal globin chain synthesis. Thalassemia is characterized by reduced rate of production (synthesis) of normal Hb due to absence or decrease in the synthesis of one or more types of globin polypeptide chains.[2] Clinically, these disorders are known as the thalassemia syndromes, resulting from both the underproduction of Hb and imbalanced globin chain synthesis, leading to a shortened red cell survival rate. The two principal types of thalassemia are alpha (α) and beta-thalassemia. They arise due to the reduced rate of synthesis of the corresponding α-chain and β-chain. The prevalence of beta-thalassemia trait and sickle cell in various regions of India is around 3%–17% and 1%–44%, respectively, because of consanguinity, caste, and area endogamy.[3] Every year, around ten thousand children with beta-thalassemia major are born in India, which constitutes about 10% of the total global load of beta-thalassemia.[2] The clinical spectrum of these disorders varies from asymptomatic conditions (beta-thalassemia minor) to serious disorders such as thalassemia major that require regular blood transfusions and extensive medical care. Accurate and timely detection of various known and unknown Hb variants can prevent the occurrence of serious Hb disorders such as thalassemia major in the newborns.[3]
Cation exchange high-performance liquid chromatography (CE-HPLC) is one of the best methods for screening, detection, and identification of various hemoglobinopathies with rapid, reproducible, and precise results. It has the advantage of quantifying Hb F and Hb A2 along with Hb variant screening in single and highly reproducible system. The simplicity of the automated system with internal sample preparation, superior resolution, rapid assay time, and accurate quantification of Hb fractions establishes CE-HPLC as an ideal methodology for routine clinical laboratory. Definite identification of mutations, specifically in case of rare variants of hemoglobins, can be achieved by molecular methodologies. The objectives of our study were to determine the prevalence of various hemoglobinopathies in our study population and to characterize each of the detected hemoglobinopathy. In addition, whenever possible, our study also aimed to identify any rare Hb variant by DNA sequencing.
Materials and Methods
The present study with 65,779 cases for hemoglobin variant analysis was carried out at Central Reference Laboratory, Metropolis Healthcare, Mumbai, India, from 2011 to 2015. The number of male and female cases in our study was 16,133 (24.53%) and 41,624 (63.28%), respectively. The number of pediatric cases accounted for 8022 (12.19%) entries. Patients who came for a premarital checkup voluntarily, patients with abnormal hemograms suggestive of hemolytic anemia, and those patients who had a positive family history of thalassemia were included in the study. Fresh whole blood (2 ml) from all the study subjects was collected in tube containing dipotassium ethylenediaminetetraacetic acid (EDTA) (Becton Dickinson Vacutainer Systems) and used as clinical specimen in this study. All of the EDTA samples involved in the study were collected from peripheral centers of Metropolis Healthcare Ltd. All the study samples were processed in the central reference laboratory, Mumbai. These samples were stored at ambient temperature, transported to the laboratory, and analyzed within 24 h after collection. Every specimen was subjected to complete blood count (CBC) and then analyzed on the Bio-Rad Variant II CE-HPLC system with use of the Variant II-thalassemia short program (Bio-Rad Laboratories) as described in the instruction manual for the assay. Concentration of Hb (%) was calculated by determining the area of a peak as a fraction of the total area of all Hb peaks seen on the CE-HPLC chromatogram [Figure 1 and Table 1]. The retention time was measured in minutes. The integrated peaks were assigned to manufacturer-defined windows derived from the retention time, i.e., the time in minutes from sample injection to the maximum point of the elution peak, of normal hemoglobin fractions and common variants. Patients with recent history of transfusion (457 cases), i.e., within 3 months before sample collection, were excluded from the study. In the current study, the samples were not collected for the purpose of the study. Our study focused only on re-analyzing the available data. In fact, our study did not perform recollection of the samples from the study participants. The data were retrieved only from the archived samples and it was intended solely for the research purpose. This was a retrospective study that involved data analysis. Since this study was not planned before sample collection, consent of the study participants was not obtained. However, the permission regarding the same was sought from the independent Ethics Committee.
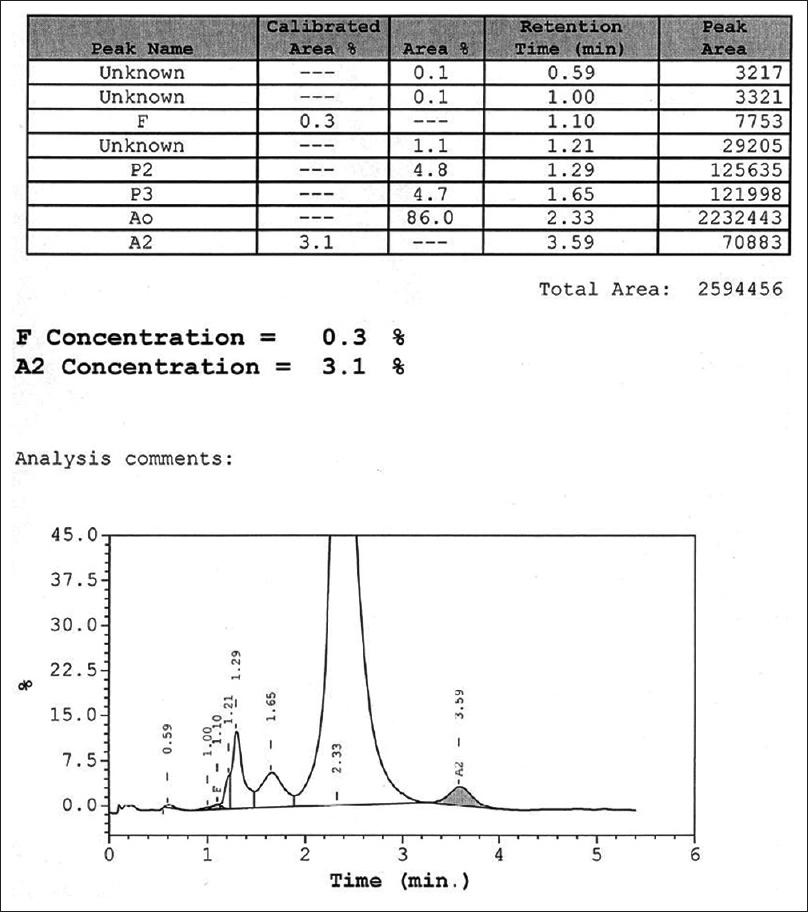
- The standard chromatogram of high-performance liquid chromatography
Some cases that could not be distinguished by CE-HPLC were forwarded for molecular characterization and identification. We sequenced the entire beta-globin gene to identify the mutations in exons 1–3 and splice site junctions. Polymerase chain reaction amplification of β-globin gene was done using primer pairs described by old[4] and then followed by bidirectional sequencing. The sequenced products were electrophoresed on DNA sequencer (3500Dx Genetic Analyzer; Applied Biosystems) and further analyzed. HbVar database (URL: http://globin.cse.psu.edu/globin/hbvar/) of human Hb variants and thalassemia mutations was referred for identification of genetic variants.
Results
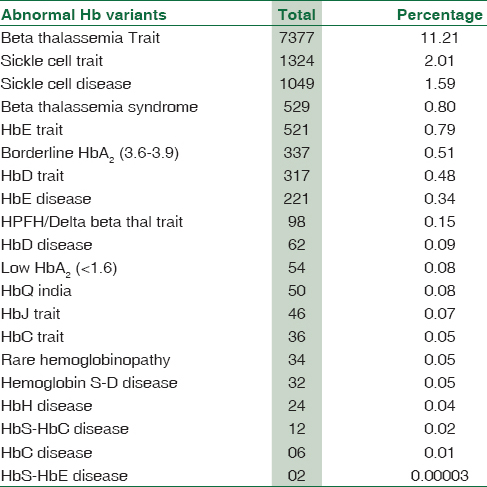
Our study recorded 12,131 (18.44%) cases of abnormal Hb fractions and 53,183 (80.85%) cases of normal Hb on CE-HPLC, as shown in Table 2.
Out of the total cases, major abnormality of Hb was beta-thalassemia trait (BTT) (n = 7377) (11.21%). The retention time for Hb A2 ranged between 3.3 and 3.9 min.
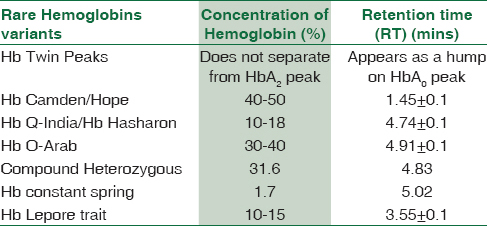
Clinically significant variants of Hb (HbS, HbE, borderline HbA2, HbD heterozygous and hereditary persistence of fetal hemoglobin [HPFH]) were found in the range of around 2% to 0.15%. Remaining variants of Hb accounted in the range from 0.09% to 0.00003%. Rare Hb variants such as Hb twin peaks (10), Hb camden/hope (12), HbQ India/Hb Hasharon (3), Hb O-Arab (2), and one case each of compound heterozygous, Hb constant spring, Hb Lepore trait, sickle cell trait (SCT) + coexist α-thal, delta beta-thalassemia homozygous, BTT + HPFH, Hb Hasharon, HbG-Philadelphia, and Hb O-Arab Hasharon were also detected. The compound heterozygous consisted of Hb S and Hb Hasharon. They all were reported based on their characteristic retention time and concentration.
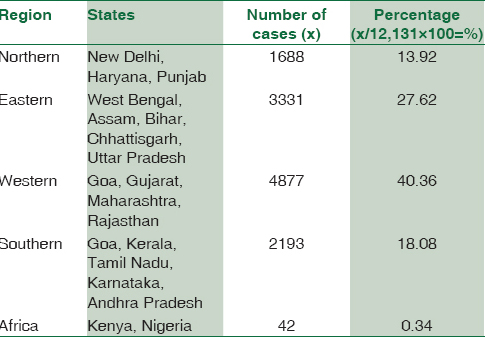
Ethnicity-wise data collection from all the study participants could not be performed during the sample collection stage since this was a retrospective study. Therefore, ethnicity data were not included in our paper. However, the data were segregated based on geographical locations, which is described in Table 3.
We considered CBC and peripheral smear findings for the interpretation of HPLC graphs.
Discussion
Alpha- and beta-thalassemia are the most common single-gene Hb disorders in the world. These disorders, which were mainly confined to certain area, religion, cast, and tribe, are now widely prevalent worldwide. Ever increasing global human migration and marriages of both, from different communities and endogamous norm, have significantly impacted the rise in cumulative cases of hemoglobinopathies worldwide. Indian population comprises numerous casts and tribal groups, each revealing different genetic traits. In India, certain communities such as Sindhi, Gujarati, Punjabi, and Bengali are more commonly affected with beta-thalassemia (incidence varying from 1% to 17%).[5]
A study carried out by Christianson et al.[6] estimate the prevalence of pathological hemoglobinopathies in India being 1.2/1000 live births. This suggests the annual birth of 32,400 babies with a serious Hb disorder. These facts compel us in employing newer techniques for early detection, prevention, and treatment of hemoglobinopathies.
In India, the most common beta-thalassemia mutations involve IVS1-5 G→C, IVS1–1 G→T, 619 bp deletion, codon 8/9 (+G), and codon 41/42 (−TCTT). They account for over 90% of the mutations in beta-thalassemia patients.[1] Molecular diagnosis of hemoglobinopathes and thalassemia is of growing importance to confirm a provisional diagnosis, such as significant sickling disorders or beta-thalassemia major. Molecular diagnosis explains a hematologic abnormality such as anemia, microcytosis, or polycythemia. It helps identify an abnormality in the presymptomatic phase, as in neonatal screening and to predict serious disorders of globin chain synthesis in the fetus and offer the option of termination of pregnancy. Finally, molecular diagnosis permits genetic counseling of prospective parents thereby permitting preoperative screening for the presence of sickle cell hemoglobin.[1]
CE-HPLC is emerging as one of the best methods for screening, detection, and identification of several hemoglobinopathies with rapid, reproducible, and precise results.[7] In our study, we recorded 12,131 (18.44%) abnormal Hb variants and thalassemia cases. BTT comprised the largest subgroup (n = 7377) (11.21%) of abnormal Hb fractions. A cutoff of over 3.9% was taken for diagnosis of BTT. Characteristic hematological findings in a typical case of BTT include microcytosis with raised RBC counts. The retention time for HbA2 ranged between 3.3 and 3.9 min. In comparison to our study, the number of BTT cases was found to be less in the studies carried out by Sachdev et al.[8] (number of BTT cases = 8.9%) and Bhalodia et al.[9] (number of BTT cases = 5.2%) on the Indian population.
Our study recorded 337 (0.51%) cases of borderline high HbA2 (3.5%–3.9%). Conditions with borderline Hb A2 need careful evaluation and interpretation. Careful evaluation of indices with iron profile will usually help in such cases. Iron deficiency may lead a low HbA2 and hence may mask a thalassemia trait, whereas B12/folate deficiency may lead to slightly raised HbA2 leading to a false diagnosis of the trait. Similarly, some rare mutations of thalassemia or a coinheritance of delta thalassemia may lead to borderline HbA2 levels. Genetic studies should be advised in such cases for a conclusive opinion. Significance of nutritional deficiencies on the levels of HbA2 is associated with raised borderline HbA2 levels of 3.5%–3.9% as well as low borderline levels of 1.6%–1.9%. Studies suggest that iron deficiency anemia (IDA), α-triplication, and beta-thalassemia trait with silent mutations contribute to borderline HbA2 cases.[10] Some studies have reported significantly lower levels of Hb A2 by the patients of IDA, associated with α-thalassemia or δ-thalassemia. Our study observed 54 (0.08%) such cases of low HbA2 where iron studies were advised and reevaluation was asked for posthematinic therapy. Since the DNA specimen of these cases was not available, molecular investigations could not be performed.
Beta-thalassemia syndrome (BTS) comprises beta-thalassemia major and beta-thalassemia intermedia. It is a worldwide disease, but more prevalent in the Mediterranean region, the Middle East, Asian Subcontinent, South-East Asia, South-West Europe, and Central Africa.[11] It is caused by a mutation in the beta-globin gene (HBB) on chromosome number-11. Our study recorded 529 (0.8%) BTS (major/intermedia) cases. All the cases diagnosed with BTS had variable degree of anemia with hemolytic blood picture. Children with BTS tend to present characteristic symptoms within 2 years of life and are dependent on regular blood transfusions.[12]
Along with the beta-thalassemia, we also recorded the following Hb variants: HbS, HbE, and HbD.
Sickle cell disease (SCD) is a protean disorder with incidence range in India, varying within various communities from 1% to 44%.[1] It leads to hemolytic anemia, acute vaso-occlusion, and organ damage due to recurrent erythrocyte sickling. Our CE-HPLC results include 1049 (1.59%) cases of SCD (SCD/SC-beta-thalassemia) and 1324 (2.01%) cases of SCT. All the cases with SCT had elution of variant S and the retention time from 4.3 to 4.7 min.
Hb E is the most common Hb variant in South-East Asia and the second most prevalent variant worldwide.[13] Typical clinical features include anemia, microcytosis, and hypochromia with target cells. HbE heterozygous individuals are clinically normal with HbE levels usually <35%. In our study, HbE variant included HbE disease (homozygous HB E/HB E-beta-thalassemia) accounting 221 (0.34%) cases and HbE heterozygous trait totaling 521 (0.79%) cases. HbE variant results from a beta chain mutation (β26 Glu→Lys) and tends to elute in A2 window within the retention time ranging from 3.3 to 3.9 min. Two cases of HbS-E disease were also recorded in our study.
Homozygous HbD/HbD-beta-thalassemia and heterozygous HbD accounted for 62 (0.09%) and 317 (0.48%) cases, respectively. HbD is usually seen in northwest regions of India. Homozygous HbD is rare and presents as a relatively milder form of disease whereas HbD-beta-thalassemia is more common and more serious. Double heterozygosity of HbD with HbS leads to anemia which is moderately severe form of disorder. Our study found thirty-two cases with HbSD (HbSD) disease.
HbC variant resulting from a separate single-base pair mutation in the β-globin gene causes a hemolytic anemia of mild to moderate severity in the homozygous form. Our study registered 6 homozygous and 36 heterozygous cases of HbC. All the HbC-related cases received in our laboratory were patients from African origin (Kenya/Nigeria). Heterozygotes are not symptomatically affected. All the HbC cases identified in our study were in African people only, who are either migrated to India or native African people. We have received such samples for confirmation, from African countries. Moreover, our study is about the prevalence of hemoglobinopathies in a tertiary care reference laboratory in India. Double heterozygosity for HbC and HbS leads to a severe sickling disorder. Twelve cases (0.02%) of double heterozygous HbS-HbC disease were reported in our study.
The three α-gene deletion (--/-α) manifests in the HbH disease. Nondeletion HbH disease is clinically more severe form than the typical deletion HbH disease. HbH dimer or α-thalassemia cannot be screened by short beta-thalassemia program. The M-shaped peak is eluted before 1 min, representing HbH and Hb barts. The CE-HPLC chromatogram as shown in Figure 2 shows the presence of sharp bands close to the injection point. HbH disease was rechecked by capillary electrophoresis. Our study registered 24 (0.04%) cases and reported M-shaped peaks that occurred at <1 min were suggestive of HbH disease.
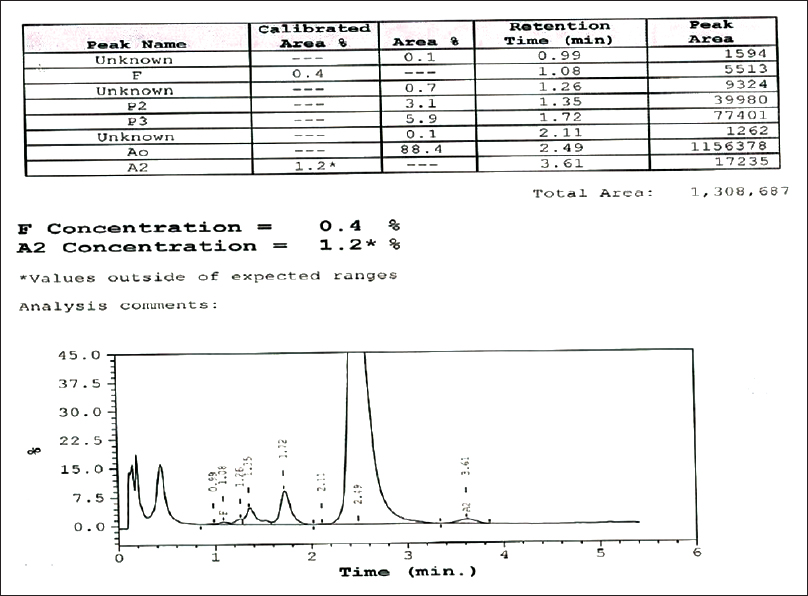
- Cation exchange high-performance liquid chromatography chromatogram of the patient's blood sample showing hemoglobin H beside the normal hemoglobin (hemoglobin A2 and hemoglobin F)
HPFH was first documented in Ghana[7] and has also been described in non-African populations. In HPFH, expression of the gamma-globin gene of Hb F persists at high levels in adult erythroid cells. Our study reported 98 (0.15%) cases of HPFH.
HbO-Arab (two cases in our study) results from point mutations of the β-globin gene. Although both heterozygous and rare homozygous individuals may be diagnosed on the basis of CE-HPLC and electrophoretic analysis, clinical manifestations are minimal. The major clinical impact of these hemoglobinopathies results from coinheritance with HbS.[14]
Majority of HbJ (α-chain variant) group of hemoglobinopathies present as an elevated P3 peak on CE-HPLC. It is important to note that a P3 peak up to six percent is usually considered normal. Our study reported 46 (0.07) cases of HbJ. Values of 6% to 12% may indicate suboptimal specimen. HbJ is usually asymptomatic or patients may present with mild anemia.
HbQ-India constituted 50 (0.08%) cases. Hb Q-India is a rare alpha chain structural variant caused by the mutation AAG→GAG (Asp→His) in the position of codon 64 of the alpha-1 gene. The characteristic findings include an unknown peak (range 10%–18%) on CE-HPLC with a typical retention time of 4.74 plus/minus 0.10 min.
Hb Lepore (a δβ variant) constituted only a single case. Hb Lepore elutes in A2 window with concentration of 10%–15%. Hb Lepore shows a characteristic hump on the downward slope in comparison to Hb E.
The present study also recorded three cases from neonate age group where variant Hb was eluted in very low concentration. The concentration of variant HbS was 8% and 9% in neonate case-1 and neonate case-2, respectively, while the concentration of HbD was 12.8% in the third neonate case. In view of patients being of neonatal age group with high HbF concentration, further analysis with molecular method was advised for accurate typing of case.
The HPLC chromatogram of eight cases did not provide conclusive evidence of any known hemoglobin variant. Of eight cases, six eluted in an unknown window while remaining two cases exhibited abnormally high concentrations of hemoglobin concentrations [Table 4]. Therefore, these eight cases were subjected for molecular analysis by β-globin gene sequencing. The results of DNA sequencing are stated in Table 4.
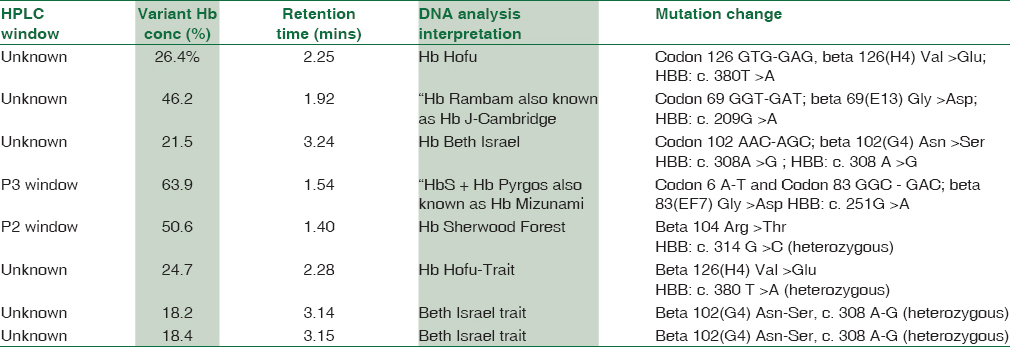
Indian studies report that prenatal diagnosis remains the most appropriate intervention to prevent the birth of homozygous children. Prenatal screening for hemoglobinopathy identifies asymptomatic individuals whose offsprings are at risk of an inherited hemoglobinopathy. This information would offer parents to undertake reproductive choices, and the pregnancy of nonimmune hydrops fetalis can also be potentially avoided. Screening can be further applied to pregnant woman/individuals from a high-risk ethnic background and consanguineous marriages. The couple at risk should be counseled regarding the nature of the disease and the implications of being carriers.[15]
Conclusion
This is the first report of a large retrospective study (65,779 cases) on the use of CE-HPLC for screening, detection, and identification of Hb variants within a clinical laboratory setting of India. The data obtained from our study find CE-HPLC (beta-thalassemia short program) as an excellent diagnostic tool for screening and detection of Hb variants and identification of unknown Hb variants. Our study recorded 7377 cases (11.21% of the total abnormal Hb CE-HPLC fractions) of BTT. This high incidence of BTT in the Indian subcontinent makes CE-HPLC-based Hb variant analysis imperative. Moreover, it also emphasizes the need for prenatal screening for prevention of clinically harmful hemoglobinopathies in offspring. Detection of other variants becomes important due to complex interactions in cases with double heterozygous and homozygous states, which may lead to severe hematological abnormalities. CE-HPLC findings must be supplemented by hemogram, family/sibling studies, and molecular studies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to acknowledge the support provided by the management of Metropolis Healthcare Ltd., Mumbai.
References
- The genetic burden of hemoglobinopathies with special reference to community health in India and the challenges ahead. Indian J Hematol Blood Transfus. 2002;20:2-7.
- [Google Scholar]
- Prevalence of common hemoglobinopathies in Gujarat: An analysis of a large population screening programme. Natl J Community Med. 2012;3:112-6.
- [Google Scholar]
- Prevalence of thalassemia and hemoglobinopathy in eastern India: A 10-year high-performance liquid chromatography study of 119,336 cases. Asian J Transfus Sci. 2016;10:105-10.
- [Google Scholar]
- A multi-center study in order to further define the molecular basis of beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2001;25:397-407.
- [Google Scholar]
- Molecular genetic testing of beta-thalassemia patients of Indian origin and a novel 8-bp deletion mutation at codons 36/37/38/39. Genet Test. 2003;7:163-8.
- [Google Scholar]
- March Dimes Global of Report on Birth Defects: The Hidden Toll of Dying and Disabled Children. March of Dimes Birth Defects Foundation 2006
- [Google Scholar]
- Advantages in the use of high performance liquid chromatography technique for screening hemoglobinopathies in Venezuela. Invest Clin. 2004;45:309-15.
- [Google Scholar]
- Detection of Hb variants and hemoglobinopathies in Indian population using HPLC: Report of 2600 cases. Indian J Pathol Microbiol. 2010;53:57-62.
- [Google Scholar]
- Study of hemoglobinopathies in patients of anemia using high performance liquid chromatography (HPLC) in Western India. Natl J Community Med. 2015;6:35-40.
- [Google Scholar]
- Significance of borderline hemoglobin A2 values in an Italian population with a high prevalence of beta-thalassemia. Haematologica. 2008;93:1380-4.
- [Google Scholar]
- Discrimination of beta-thalassemia minor and iron deficiency anemia by screening test for blood cell indices. Turk J Med Sci. 2012;42:275-80.
- [Google Scholar]
- The burden of hemoglobinopathies in India and the challenges ahead. Curr Sci. 2000;79:1536-47.
- [Google Scholar]
- Hemoglobin E disease in North Indian population: A report of 11 cases. Hematology. 2007;12:343-7.
- [Google Scholar]
- Laboratory investigation of hemoglobinopathies and thalassemias: Review and update. Clin Chem. 2000;46(8 Pt 2):1284-90.
- [Google Scholar]
- Microcytic hypochromic anemia: Should high performance liquid chromatography be used routinely for screening anemic and antenatal patients? Indian J Pathol Microbiol. 2013;56:109-13.
- [Google Scholar]



